Cells, media, and reagents
RAW264.7, a macrophage cell line, was cultured in Dulbecco’s minimum essential medium (DMEM; HyClone, Logan, USA) added with 10% fetal bovine serum (FBS; Gibco, New York, USA), 100 U/ml penicillin (Gibco, New York, USA), and 100 μg/ml streptomycin (Gibco, New York, USA). ATDC5, a chondrogenic cell line, was cultured in DMEM: nutrient mixture F12 (DMEM: F12; HyClone, Logan, USA) supplemented with 5% FBS. The cells were placed at 37 °C under humidified conditions with 5% CO2. The bacterial LPS of Escherichia coli and the recombinant mouse IFN-γ and IL-4 were purchased from PeproTech (Rocky Hill, USA). Exosome-depleted FBS was bought from System Biosciences (California, USA).
Induction of macrophage polarization
RAW264.7 cells were seeded on culture plates, incubated at 37 °C overnight to adhere to the bottom of the plates. Cells were stimulated with a complete medium containing 100 ng/ml LPS plus 20 ng/ml IFN-γ 24 h to induce M1 polarization and 20 ng/ml IL-4 24 h to stimulate M2 polarization [6].
Exosome isolation and identification
RAW264.7 cells were induced to an M1/M2 type for 24 h and cultured in an exosome-depleted FBS-containing complete medium. The supernatant was collected, and exosomes were obtained through ultracentrifugation [35]. Briefly, the supernatant was subjected to a series of differential centrifugation steps (300×g for 10 min, 2000×g for 10 min, and 10,000×g for 30 min) to remove intact cells and cell debris. Subsequently, the supernatant was centrifuged at 100,000×g at 4 °C for 70 min to isolate the proteins-containing exosomes. The exosomes were then purified by washing them with PBS and subjected to an additional centrifugation step at 100,000×g for 70 min.
The protein content of the exosomes was measured with a bicinchoninic acid (BCA) protein assay kit (Beyotime Biotechnology, Shanghai, China) referring to the manufacturer’s instructions. CD9, CD63, CD81, TSG101 and Calnexin (Proteintech, Chicago, USA), which were exosomal marker proteins, were assessed by western blotting as previously described [51]. M2 macrophage-derived exosomes were scanned using a TEM (JEM1400, Tokyo, Japan). The size and concentration of exosomes were analyzed through NTA (Nanosight NS300, Malvern, UK).
Exosome labeling and cellular uptake
M2 macrophage-derived exosomes were labeled with fluorescent dyes referring to a previous study [52]. They were labeled using a PKH26 red fluorescent cell linker kit (Sigma-Aldrich, St. Louis, USA) in accordance with the manufacturer’s instructions. The labeled exosomes were washed and centrifuged at 100,000×g for 60 min. Then, the resuspended exosomes were deployed to RAW264.7 and ATDC5 cells for 6 h. After the supernatant was removed, FITC-conjugated phalloidin (Sigma-Aldrich, USA, St. Louis, USA) and 4,6-diamidino-2-phenylindole (DAPI; Beyotime Biotechnology, Shanghai, China) were used to stain the cytoskeleton and nuclei, respectively. The cells were washed with PBS twice and photographed with a confocal microscope (Leica TCS-SP5, Germany).
Exosomal miRNA sequencing and analysis
High-throughput miRNA sequencing and subsequent bioinformatic analysis were performed by CloudSeq Biotech Co., Ltd. (Shanghai, China). The total RNA of exosomes was prepared and quantified using a NanoDrop ND-100 (Thermo Fisher Scientific, USA). Then, total RNA of each sample was used to prepare the miRNA sequencing library. The libraries were denatured as single-stranded DNA molecules, captured on Illumina flow cells, amplified in situ as clusters, and sequenced for 50 cycles on an Illumina HiSeq4000 sequencer (Illumina, CA, USA). Raw data were generated through sequencing, image analysis, base calling, and quality filtering by using an Illumina sequencer.
RNA extraction, reverse transcription-PCR and real-time qPCR
The total RNA of cells was extracted using TRIzol reagent (Invitrogen, CA, USA), and NanoDrop ND-100 was used to measure the quality and concentrations of RNA. cDNA was synthesized with TaqMan reverse transcription reagents (Applied Biosystems, CA, USA). Real-time qPCR was performed using an ABI 7500 system (Applied Biosystems, CA, USA) in accordance with previously described methods [53]. The reverse transcription primers of miRNAs were prepared using the stem-loop method. The transcript levels of mRNA and miRNA were normalized to β-actin and U6, respectively. Gene expression levels were analyzed using 2−ΔΔCt method. The primers used for these analyses are listed in Additional file 1: Table S1.
Lentivirus preparation and construction of stably transfected cell lines
pHBLV-zsgreen-puro (miR-ctrl) and pHBLV-miR-26b-5p-zsgreen-puro (miR-26b-5p overexpression) plasmids were prepared by Hanbio Biotechnology Co., Ltd. (Shanghai, China). The prepared plasmids were transfected into HEK293T cells with LipoFiter™ 3.0 (liposomal transfection reagent). The virus supernatant was collected and filtered using a 0.45 μm cell strainer 48 h after transfection. The supernatant was further centrifuged to 72,000 g at 4 °C for 120 min and resuspended with fresh media for the following experiments. The titer of the concentrated lentivirus was measured reaching 2 × 108 TU/ml.
RAW264.7 and ATDC5 cells were seeded in six-well plates (5 × 105/well) and incubated overnight. Then, 6 μg/ml polybrene (Sigma-Aldrich, St. Louis, USA) was applied before transfection. The lentivirus-containing medium was changed with complete media 24 h after transfection, and fluorescence signal was observed using a fluorescent microscope 48 h after transfection. Green fluorescence indicated successful transfection in cells. Then, 4 μg/ml puromycin was added to the culture medium to kill untransfected cells. After about 7 days, the successfully transfected cells survived, and they were used for the following analysis.
Dual-luciferase construction and reporter assay
pmirGLO dual-luciferase miRNA target expression vectors were prepared by GenePharma (Suzhou, China) to assess the miRNA activity. The 3ʹ UTR sequences of TLR3 and COL10A1 (wild type or mutant) were cloned in the vectors. The firefly luciferase reporter gene was controlled by an SV40 promoter, and Renilla luciferase was used as a control reporter for normalization. HEK293T cells with overexpressed miR-ctrl or miR-26b-5p were co‐transfected with various pmirGLO vectors for 24 h. Then, the luciferase activity in the lysates was assessed with a dual-luciferase reporter gene assay kit (Beyotime Biotechnology, Shanghai, China) in accordance with the manufacturer’s instruction.
Cell co-culture experiments
RAW264.7 cells were polarized to an M1 type by stimulating with LPS and IFN-γ for 24 h. M1 macrophage supernatants were centrifugated at 1000×g for 5 min and diluted with a serum-free medium (1:1) to prepare M1-CM for the following experiments. The obtained M1-CM was deployed to RAW264.7 to evaluate the effects on macrophage polarization and added with insulin, transferrin, and selenous acid (ITS; Sigma-Aldrich, St. Louis, USA) to evaluate the chondrogenesis of ATDC5 cells [39]. M1-CM was analyzed with a Luminex liquid chip for multi-cytokine detection to assess the effects of miR-26b-5p on macrophage repolarization and chondrocyte hypertrophy.
Immunofluorescence staining
RAW264.7 cells with or without miR-26b-5p overexpression were stimulated with M1-CM for 24 h. They were fixed in 4% paraformaldehyde containing 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, USA), and 1% bovine serum albumin (BSA) was applied to block nonspecific binding. Afterward, the cells were incubated with antibodies, including CD16/32 (BD, CA, USA), CD206 (BD, CA, USA), and TLR3 (Abcam, Cambridge, MA, USA), overnight. They were further incubated with fluorescent secondary antibodies (Abcam, Alexa Fluor 594, Cambridge, MA, USA) and DAPI were transferred to cells and observed using a confocal fluorescence microscope (Leica TCS-SP5, Germany).
Flow cytometry analysis of macrophage subsets
CD16/32 and CD 206 were chosen to respectively mark M1 and M2 phenotypes and to distinguish M1 and M2 macrophage populations. After being washed thrice with PBS, the polarized macrophages were digested and resuspended with PBS. Then, 5 μl of Alexa anti-CD206 (Fluor 647-conjugated, BD, CA, USA) and 5 μl of anti-CD16/32 (PE-conjugated, BD, CA, USA) were applied to evaluate the macrophage subsets. Alexa Fluor 647 isotype control (BD, CA, USA) and PE isotype control (BD, CA, USA) were transferred to exclude the cells with nonspecific staining. M1 macrophages were regarded as CD16/32-positive and CD206-negative cells, while M2 macrophages were denoted as CD206-positive and CD16/32-negative cells.
Western blotting analysis
The cells were washed with PBS and lysed with a radioimmunoprecipitation assay buffer (RIPA; Millipore, MA, USA) containing protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, USA). The protein concentrations of cells were evaluated using a BCA protein assay kit. Then, 20 μg of proteins was separated through 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk for 2 h and incubated with primary antibodies overnight. The membranes were further incubated with fluorescent secondary antibodies and washed thrice with Tris-buffered saline with Tween (TBST). Fluorescent signals were photographed with LI-COR imaging systems (Lincoln, USA).
Alizarin red staining
The chondrogenesis of ATDC5 cells was induced for 14 days. Then, the cells were fixed with 4% paraformaldehyde, incubated with 1% Alizarin red solution (Sigma-Aldrich, St. Louis, USA) for 30 min, [39] and washed with PBS twice to remove excess Alizarin red dye. Afterward, they were photographed under an optical microscope.
Establishment of ACLT-induced OA mouse model
Animal experiments were approved by the ethics committee of the First Affiliated Hospital of Soochow University. Twenty-four 3-month-old male C57BL/6J mice obtained from Laboratory Animal Center of Soochow University were exposed under specific pathogen-free (SPF) conditions and fed with commercial food and water. OA was induced by applying ACLT, which can cause abnormal mechanical loading, to the right knee in accordance with previously described methods [54, 55]. An anterior drawer test was conducted to confirm complete transection, and a sham operation was made by opening the joint capsule and suturing the incision.
The mice were randomly and averagely split into four groups and assigned to four cages. The mice in group 1 were assigned in the control group and treated with sham operation. The mice in groups 2–4 were treated with ACLT to the right knee joints. The mice in group 2, 3, and 4 were also intra-articularly injected with PBS, 5 nmol agomir NC, and 5 nmol miR-26b-5p agomir, respectively. Intra-articular injection was administered once a week for 4 weeks [45, 46].
Gait analysis
Gait analysis was performed using a gait analysis system as described previously [47]. In brief, the gait analysis system contains a fluorescent light tube along a glass plate runway. The mice were placed individually on the runway and allowed to walk across the runway freely. The gait of each mouse was recorded using a high-speed color camera and analyzed with the VisuGait software (Xinruan Co., Ltd., Shanghai, China). The swing phase duration (s) indicated the period when the paw was not touching the ground in a complete step cycle. The swing speed (m/s) was calculated by dividing the stride length by the duration of the swing phase. The duty cycle (%) was defined as the ratio between stance duration and complete step cycle duration [56]. The ratios of the affected right hind limb to the contralateral left hind limb (RH/LH) were calculated to eliminate individual differences [50].
Von Frey withdrawal threshold testing
Von Frey filaments were applied to assess the secondary mechanical allodynia by measuring the withdrawal threshold in accordance with previously described methods [56]. The mice were placed in a transparent plexiglass chamber with a metal mesh floor for at least 10 min to acclimatize them before the test. The filaments were applied when the mouse stood still on all four paws and operated thrice with an inter-trial interval of 10 s. The response was considered positive when the mice flinched their paw more than once. Then, the paw withdrawal threshold (PWT) of the mouse was recorded.
Histological observation
The mice were killed 4 weeks after the injection. The right knee joints of the mice in various groups were dissected and fixed with 4% paraformaldehyde for 48 h. The samples were decalcified in 10% EDTA for 3 weeks and embedded in paraffin. The sagittal sections of the medial compartment of the knee joints were cut at 4 μm for the following microstructural observations. They were further stained with S&F and H&E. The OARSI score was applied in accordance with previously described methods [57]. Synovitis was assessed using the Krenn scoring system [58]. Immunohistochemical staining was applied in accordance with a previous report [59]. Specifically, primary antibodies, including CD16/32, CD206, COL10, and MMP-13 (Abcam, Cambridge, MA, USA, dilution 1:200) were applied and incubated at 4 °C overnight. A secondary antibody was incubated at 25 °C for 1 h. The samples were stained with diaminobenzene (Dako, North Sydney, NSW, Australia) and hematoxylin (Sigma-Aldrich, St. Louis, USA). The immunohistochemically stained samples were then photographed under a microscope. The percentage of positively stained cells in the articular cartilage and synovium was calculated (Additional file 1).
Statistical analysis
Data were expressed as mean ± standard deviation. Differences between two groups were compared via two-sided Student’s t-test. Multifactorial comparisons were performed through one-way analysis of variance. Data with p < 0.05 were considered to have statistically significant differences. In this study, “*”and “**” denoted p < 0.05 and p < 0.01, respectively. The data analysis was calculated with SPSS 22.0 analysis software (SPSS Inc, Chicago, IL, USA).




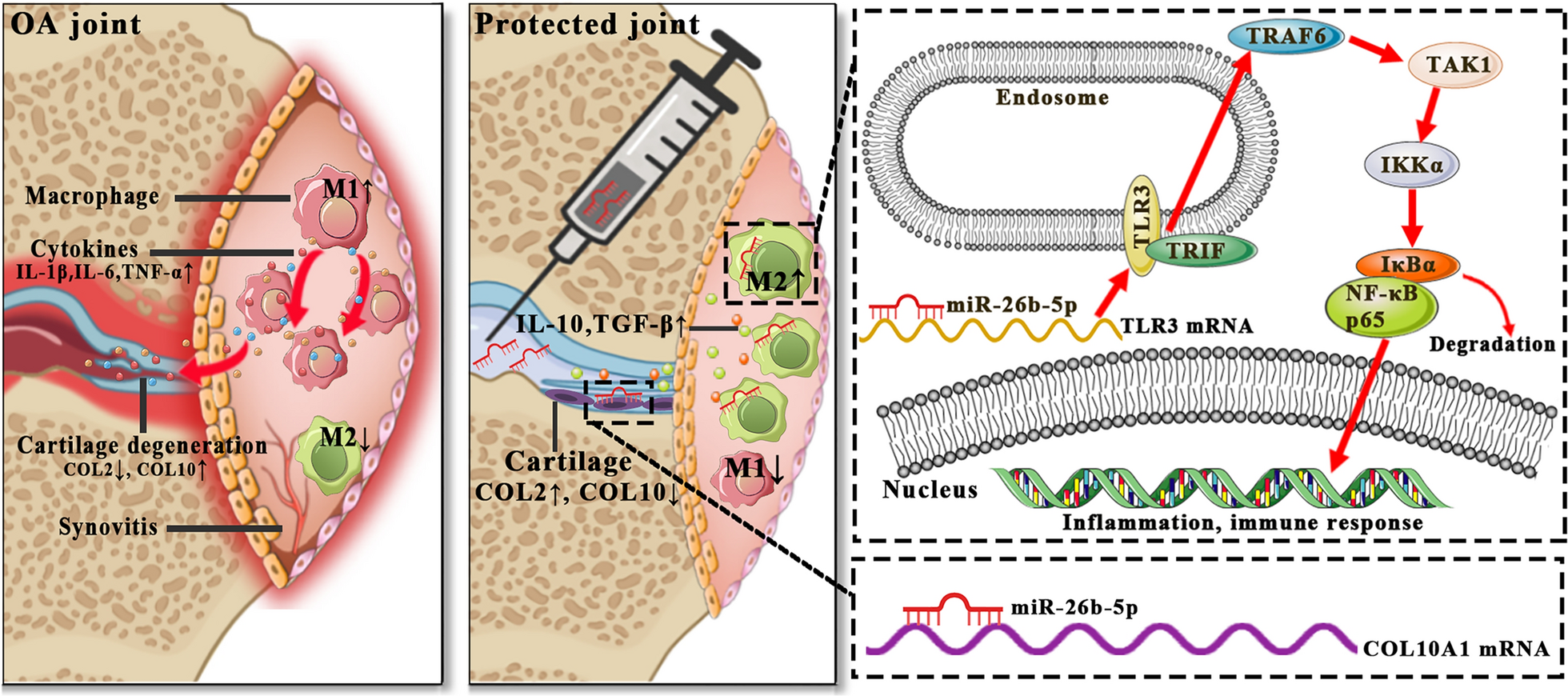

Comments (0)