
Remember me

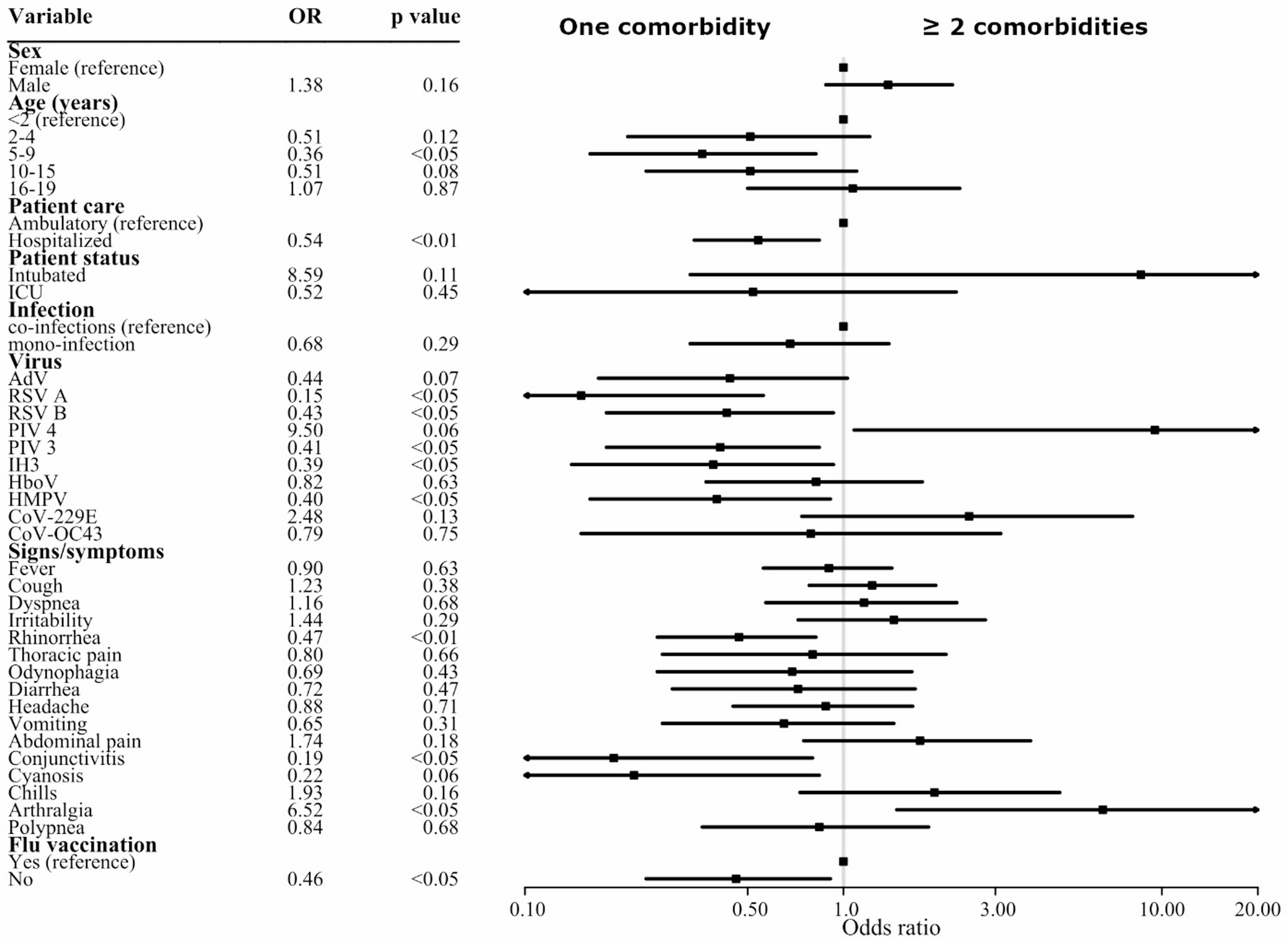
Twenty-seven participants (68% of planned enrolments) were allocated to one of three groups: control (HC, number of participants, n = 10), intermittent fluconazole user (IF, n = 10) and maintenance fluconazole user (MF, n = 7). Participant baseline characteristics are displayed in Table 2. The average age of participants was 31 (range: 18 to 41) years. No significant differences in ethnicity, menstrual cycle length, sexual activity, medication use or pregnancy history were observed between groups (Table 2, S2-S3, all p > 0.05). The average measure of vaginal pH was 5.0 +/- 0.4 with no significant differences observed between groups (Table S3, p = 0.49). Participants in the IF treatment group primarily took 150 mg as a single dose as required for symptomatic episodes (n = 9), while one participant required 50 mg three times in a week for their symptomatic recurrence (Table S1). Participants in the MF treatment group mostly took 50 mg of fluconazole daily (n = 4, Table S1). All participants in the MF group had used fluconazole in the four days leading up to their sample collection.
Table 2 Participant characteristics of the healthy control (HC), intermittent fluconazole (IF) and maintenance fluconazole (MF) groupsContinuous data are expressed as mean and SD (standard deviation), and categorical data as n/N (%) (number of participants in grouping, [n] over total number of participants [N]). Significant differences were tested using Fisher’s Exact Test for categorical data and ANOVA for continuous variables.
Characterisation of the interkingdom vaginal and gastrointestinal microbiomesFollowing quality filtering the dataset generated from 27 participant samples yielded 7,451,272 sequence reads. Sequence read depths varied considerably between ITS and 16 S regions. Fewer reads were identified from the ITS region compared to the 16 S region despite the use of multiple swabs and adequate DNA concentrations. This reduced sequencing depth was more pronounced in the vaginal ITS dataset, where 19 of the 27 participants displayed a read count greater than 260 and qualified them for downstream analyses. All sequence reads, were classified into 522 taxa from the vagina and 3,629 taxa from the GIT. The interkingdom VMB consisted of taxa from eleven phyla from the Fungi and Bacteria dominated by the Firmicutes (average relative abundance = 74.6%), Ascomycota (14.1%), Actinobacteria (8.2%) and Basidiomycota (3.1%) (Fig. 1, Table S5). The VMB was represented by more than 80 genera with dominance from Lactobacillus (74.4%), Candida, (10.4%) and Gardnerella (5.0%) (Fig. 1, Table S5). A Lactobacillus-dominated environment was observed ranging from 0.02 to 98.5% relative abundance in any one participant. Eleven different Lactobacillus species were identified and numerous distinct ASVs were also identified, although classified only to the level of Lactobacillus (data not shown). The most common Lactobacillus species observed were L. crispatus, L. gasseri, L. iners L. iners A-B-1 and L. jenseni (Fig. 1). Other dominant taxa (with > 5% relative abundance in any one participant) belonged to seven different species: Candida albicans, Gardnerella vaginalis, G. swidsinskii, Rhodotorula mucilaginosa, Bifidobacterium Longum subspo. Longum, Issatchenkia orientalis, Saccharomyces cerevisiae, and Malassezia globosa (Fig. 1, Table S5).
Fig. 1
Relative abundance bacterial and fungal species in the vagina of participants. Bar plot shows the relative abundance of amplicon sequence variants (ASVs) with a relative abundance greater than 0.5% found in the vagina of the 27 participants, grouped by three treatment cohorts: healthy control (HC), RVVC patient with intermittent fluconazole use (IF) and RVVC patient with maintenance fluconazole use (MF). To account for differences in sampling depth between samples, vaginal data were rarefied prior to conversion to relative abundance. Colours indicate the summed relative abundance at the level of species from an individual vaginal swab sample
The VMB profiles of the total cohort were clustered into five CST’s (Table 3). The most prevalent CST in participants was the L. crispatus dominated CST-I (40.7% of samples) followed by CST-III L. iners (25.9%) and then CST-IV non- lactobacillus dominated (11.1%). Participants with CST-II L. gasseri, and CST-V L. jensenii were both present in 7.4% of samples, while another 7.4% of CST’s were Lactobacillus-dominated by an unassigned species.
The GIMB of participants consisted of taxa from 12 phyla with dominance by the Firmicutes and Bacteroidetes and representation from the Actinomycetota, specifically in the genus, Actinomyces (Table S6). Dominant genera included Prevotella (15.4%), Bacteriodes (11.7%) and Blautia (9.2%).
Table 3 CST: Community state type frequencyDifferences in microbiome richness and diversity between treatment groupsThe Shannon index and Chao [25, 26] are methods of alpha diversity that are commonly used to characterise the richness and evenness of the microbial taxa present in a sample. Comparatively, beta diversity is a measure that indicates the similarity and differences in composition between samples. Overall, GIT samples displayed a far greater observed number of taxa and other alpha diversity metrics when compared to vaginal samples (Figure S2, Kruskal-Wallis, p-value = < 0.05). However, there were no differences in the observed number of taxa, Shannon’s or Chao diversity metrics between groups within the GIT or vagina (Fig. 2, p-value > 0.05). Vaginal samples displayed a relatively consistent trend in alpha diversity metrics across groups, whereas GIT samples trended towards an increase in alpha diversity metrics for the MF group when visually compared with the HC and IF groups, but these trends were not significant (Fig. 2). There was also no significant difference in beta diversity between groups in either vagina or GIT samples (Figure S3, Table S7).
Fig. 2
Alpha Diversity of fungal and bacterial microbiome in vagina and GIT locations with treatment for RVVC. Alpha diversity metrics showing observed number of amplicon sequence variants, Shannon and Chao diversity metrics of vagina (top) and GIT (bottom) in treatment groups indicating testing of the mean between groups with Kruskal-Wallis test
Microbial trends in the vagina microbiome in treatment groupsEight bacterial taxa were identified in all treatment groups: L. crispatus, L. iners, L. jensenii, L. gasseri, Lactobacillus spp. (consisting of > 70 unique ASVs), L. acidophilus, L. reuteri and Gardnerella vaginalis. Despite a lack of significance at the beta diversity level, subtle differences in the relative abundance of taxa were observed between the fluconazole groups (IF, MF) and the HC group. In the unmerged data set from the 16 S (bacterial) region from the vagina, L. crispatus displayed a higher average relative abundance in the IF group with 47.6% when compared to the MF (41.1%) or HC (25.5%) groups (Figure S3). L. iners also showed variability in average relative abundance between groups (MF 3.0%, IF 18.9%, and HC 8.1%). However, these differences were not significant.
In the IF and MF groups all participants that were categorised into CST-I (n = 8) displayed high average relative abundance of L. crispatus (> 90.0%), whereas in the HC group this was the case for only one of the three of the participants with CST-I(98%) (Figure S3). Overall, IF group participants showed a higher average relative abundance of the L. iners dominated CST-III (94%) in comparison to other groups (HC 80%, MF 81%) (Figure S3).
Different bacterial and fungal taxa were observed in the VMB across all three participant treatment groups, but only two vaginal fungal species were observed consistently in all groups, they were: Candida albicans and Malassezia globosa. Moreover, Saccharomyces cerevisiae was not identified in the VMB of the MF group and Rhodotorula mucilaginosa was not identified in the HC group.
Overall, Candida spp. vaginal carriage was identified in nine participants HC (n = 1), IF (n = 6), MF (n = 2). All participants displaying vaginal Candida spp. belonged to either bacterial CST-I or CST-III (n = 8) (Fig. 3a). Candida albicans dominated among all Candida spp. in 95% of all Candida spp. positive samples in the vaginal environment. Other non- albicans Candida (NAC) species accounted for the remaining 5%.
A significant difference in Candida spp. VMB abundance was observed between the IF and HC groups (p = 0.025), with a greater average, yet non-significant, relative abundance in the IF and MF groups (p = 0.38) (Fig. 3b). Candida albicans was the most dominant Candida spp. observed. In interkingdom analysis four vaginal samples showed a higher relative abundance of C. albicans than their dominant CST associated Lactobacillus spp. (IF, n = 2, MF, n = 1, HC, n = 1, Fig. 1).
Fig. 3
a Candida spp. relative abundances classified via CST group. Data displays relative abundance of Candida spp. in the vagina of participants depending on their Community State Type classification. Data were merged between ITS and 16 S sites from vaginal samples. Testing of the mean relative abundance differences between groups were conducted with Kruskal-Wallis one way analysis of variance. Data were rarefied prior to plotting. b Candida spp. relative abundances in the vagina. Data displays relative abundance of Candida spp. in the vagina of participants depending on their treatment group (IF, MF or HC). Data were merged between ITS and 16S sites from vaginal samples. Testing of the mean relative abundance differences between groups were conducted with Kruskal-Wallis one way analysis of variance. Data were rarefied prior to plotting
Specific bacterial and fungal species were assessed for interkingdom between-group differences. The between group difference of the vaginal bacterium G. vaginalis was significant, with an elevated abundance observed in the HC group compared to the IF and MF groups (Kruskal Wallis p < 0.05) (Fig. 4a). A significantly greater average relative abundance of vaginal R. mucilanigosa (microfungus) was observed in the IF group compared to both the HC and MF groups (p < 0.05) (Fig. 4b).
Fig. 4
Effect of treatment on dominant species in the vaginal microbiome. Data displays relative abundance of selected relevant taxa in the vagina of participants grouped by treatment. Horizontal lines show different strains of the same species represented in the cohort 4a) G. vaginalis, 4b) R. mucilanigosa. Testing of the mean relative abundance differences between groups were conducted with Kruskal-Wallis one way analysis of variance. Data were rarefied prior to plotting
Candida albicans in the gastrointestinal microbiome siteCandida albicans was also the dominant Candida spp. in the GIT environment (Table S6).
Compared to other groups, the IF group showed no significant dominance of C. albicans in the GIMB and the HC and MF groups had very low GIMB abundance of any Candida species (Table S6).
GIT samples were analysed for differences in the abundance of Firmicutes and Bacteroidetes and the MF group displayed a lower average relative abundance of Bacteroidetes compared with the HC and IF groups (Figure S5).
Vaginal and GIT Candida pairing and dual site Candida carriage (co-carriage)Dual site carriage of Candida, where both the GIT and vagina sites have an occurrence of Candida spp., was assessed. Five participants (18.5%) of the overall sample recorded dual carriage of Candida spp. All of these samples originated from fluconazole users (IF = 3, MF = 2) (Fig. 5). Candida species were not consistently identified in both sites e.g., participants displayed GIT Candida spp. without any vaginal Candida spp. and vice versa (Fig. 5). Twelve participants (44.4%) displayed Candida spp. only in the GIT samples (HC = 7, IF = 3, MF = 2), only three (25%) of these were C. albicans. Five (18.5%) displayed no Candida spp. in either site (HC = 2, IF = 1, MF = 2) (Fig. 5).
When co-carriage was assessed for pairing e.g., the same species occurring in both the VMB and GIMB, only two participants in the IF group displayed a vaginal/GIT C. albicans pairing. When paired samples were assessed for Candida spp. vaginal abundance, abundance was higher. No NAC species were identified as being paired between sites (Fig. 5).
Two NAC species were seen predominately in the GIT environment; C. saken = 8 (HC = 3, IF = 3, MF = 2) with no vaginal carriage and C. hyderabadenis, which was predominantly found in the GIT n = 6 (HC = 3, IF = 2, MF = 1), none of these six displayed it vaginally. It was, however, observed in the vaginal sample for one IF participant (3.7%) (Fig. 5). Overall, five participants (18.5%) presented with Candida spp. only in the vagina (HC = 1, IF = 3, MF = 1).
Fig. 5
Candida carriage and pairing in gastrointestinal and vaginal microbiome. Data displays raw abundance of Candida spp. in the vagina and GIT shown per participant to demonstrate dual site relationships. Horizontal lines show a different strains of the same species. Data were normalised prior to plotting
Vaginal and GIT Lactobacillus pairing and dual site Lactobacillus carriage (co-carriage)Similar to assessing for Candida spp. co-carriage, the samples were also assessed for Lactobacillus spp. co-carriage and pairing. The majority of GIMB samples (n = 24 out of 27) did not display any Lactobacillus spp. at any site. Only three participants presented with co-carriage of Lactobacillus spp. (HC, n = 1, IF, n = 1, MF, n = 1). No pairing was observed between sites (Figure S6).
Presence of Non- Candida fungal spp. in the VaginaFungal species that did not belong to the Candida genus were identified across groups in the VMB. 90% of the IF group (n = 9, 90%) presented with fungi including Candida and non-Candida species, compared to 72% of the MF and 50% of the HC groups (Table S5).
When dominant vaginal fungal characteristics were assessed, five IF participants (50%) showed dominance of non-Candida fungi. Four MF (57%) and four HC (40%) participants displayed non-Candida fungal kingdom dominance (Table S5). In the IF treatment group participants, #211 and #212 displayed R. mucilaginosa (average relative abundance 32.4% and 38.4%) with a CST-I bacterial microbiome (average relative abundance 63.0 & 61.1%). Participant #229 displayed I. orientalis dominance (16.2%) and a small < 1% abundance of C. albicans in an CST-III (83.5%) environment. Participant #223 displayed C. albicans dominance (9.5%) and five other non-Candida spp. were observed, with M. globosa (4.6%) in a CST-I (72.3%) environment being the most abundant species. Participant #226 in the MF group displayed a diverse mycobiome of 20 different species, 19 of which were non-Candida spp. M. globosa was the most abundant (2.0%), found with a CST-I (85.5%) bacterial community. The HC group participant #218 showed S. cerevisiae dominance (41.3%) with unassigned CST L. acidophilus-dominance. Two HC group participants displayed interkingdom relative abundance of fungal microbes higher than their dominant CST bacterial community; #220 was dominated by C. albicans (47.4%) and also carried five other non-Candida spp. at low levels and a CST-I (41.2%) bacterial environment. Participant #202 displayed I. orientalis (45.3%) as the dominant fungus and bacterial CST-IV with G. vaginalis being dominant (31.0%).
Comments (0)