
Remember me

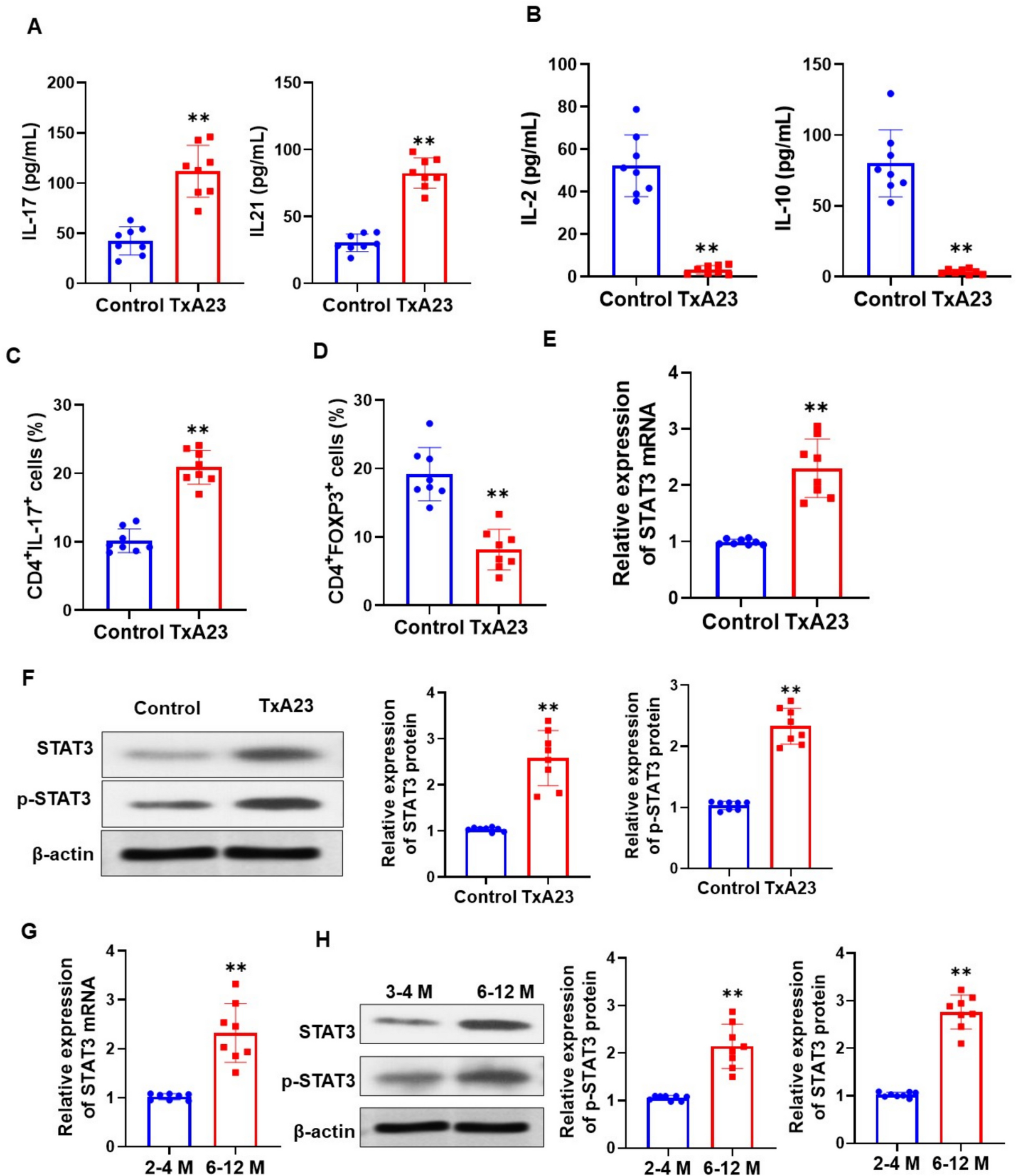


The study group consisted of 120 randomly selected juvenile patients with long-standing (over 1 year) type 1 diabetes mellitus (LS-T1D) recruited from the Clinic of Pediatrics, Department of Diabetology and Endocrinology, Medical University of Gdansk, Poland. Subjects with other autoimmune disorders, microvascular complications, acute inflammatory and infectious diseases, history of hypertension, or blood transfusions were not enrolled in the study. Type 1 diabetes mellitus (T1D) was defined in accordance with American Diabetes Association criteria.
Eight milliliters of venous peripheral blood from each individual was collected into Vacutainer tubes containing heparin (BD Bioscience, USA). The blood glucose level, biochemical measurement of renal function, lipid status, and glycosylated hemoglobin (HbA1c) were measured at the time of sampling. In all examined patients, the C-peptide levels were below 0.5 ng/ml. All patients were treated with humanized insulin. The control group consisted of 45 age and sex-matched healthy individuals with no signs of autoimmune, chronic, inflammatory, or neoplastic disease at the time of sampling and no evidence of T1D in their families.
The study followed the principles of the Declaration of Helsinki and was approved by The Ethics Committee of The Medical University of Gdansk (NKBBN/645/2019). From all parents of juvenile participants, written informed consent was obtained. A summary of the clinical characteristics of the analyzed subject groups is presented in Table 3.
Reagents and antibodiesThe following monoclonal antibodies (mAbs) were used in cytometric studies (fluorochromes and clones in brackets): anti-CD3 (PE-Cy7, SK7), anti-CD4 (APC or PE-Cy5, RPAT4), anti-CD25 (PE, 2A3), anti-CD127 (PE-Cy7, AO19D5). Items were purchased from BD Biosciences, USA, or BioLegend, USA. Intracellular staining for Foxp3 (FITC, 206D) and IFN-γ (PerCP-Cy.5.5, B27) was performed with ready-to-use kits according to the manufacturer’s suggestions (BioLegend, USA and BD Biosciences, USA, respectively). Additionally, the following reagents were used for the stimulation of cell cultures: anti-CD3 (clone UCHT1, eBioscience, USA), anti-CD28 (clone CD28.2, eBioscience, USA) antibodies, rIL-2 (BioLegend, USA), PMA (Sigma, Poland), ionomycin (Sigma, Poland), and GolgiStop (BD Biosciences, USA).
PBMC isolation and cell cultureEight milliliters of venous peripheral blood from each individual was collected in Vacutainer tubes containing heparin (BD Bioscience, USA). Peripheral blood mononuclear cells (PBMCs) were isolated from the donors’ blood by density gradient centrifugation over Histopaque (Sigma-Aldrich, Poland) and cultured on 24-well plates in RPMI 1640 (Sigma-Aldrich, Poland) supplemented with 5% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich, Poland) and 1% penicillin/streptomycin (Sigma-Aldrich, Poland).
For each study individual, the isolated PBMCs were placed in two wells. First, the control well (starting point) was designated “non-stimulated” and second “stimulated.” Cells in both wells were treated with anti-CD3 and anti-CD28 antibodies, both at a concentration of 5 µg/ml. Wells designated as “stimulated” were additionally treated with recombinant human CCL5 at a 15 ng/ml concentration. Cells in both wells were also stimulated with recombinant human IL-2 (100 U/ml). Cultures were incubated at 37 °C with 5% CO2 for 48 h and then cells were designated for cytometric analysis.
Isolation and culture of CD4 + CD25 + CD127low TregsCD4 + CD25 + CD127low Tregs were separated from PBMC by a two-step magnetic separation procedure using EasySep™ Human CD4 + CD127lowCD25 + Regulatory T Cell Isolation Kit (Stem Cell, USA). First, we used EasySep Releasable RapidSpheres along with the CD25 Positive Selection Cocktail to isolate CD25 + cells. Next, we utilized a CD4 + T-cell enrichment cocktail to ensure the exclusive isolation of CD4 + CD25 + cells. The resulting cell suspension underwent further refinement by adding CD127high Depletion Cocktail and Dextran RapidSpheres, allowing for the depletion of CD127 + cells. The final isolated fraction consisted of highly purified CD4 + CD25 + CD127low cells. The purity of isolated cells was assessed by flow cytometry. Starting with fresh peripheral blood, the CD4 + cell content of the enriched fraction was typically 94 ± 5% and the purity of selected CD4 + CD127lowCD25 + Tregs ranged from 86 to 96%.
Magnetically selected CD4 + CD25 + CD127low T cells were then cultured on the round bottom 96-well plates. For each study individual, the isolated Tregs were placed in two wells. First, the control well (starting point) was designated “non-stimulated” and second “stimulated.” Cells in both wells were treated with anti-CD3 and anti-CD28 antibodies, both at a concentration of 5 µg/ml. Wells designated as “stimulated” were additionally treated with recombinant human CCL5 at a 15 ng/ml concentration. Cells in both wells were also stimulated with recombinant human IL-2 (100 U/ml). Cultures were incubated at 37 °C with 5% CO2 for 48 h. At the same time, from each study individual, the autologous “non-stimulated” PBMCs were isolated by density gradient preparation over Histopaque-1083 (Sigma) and cultured for 48 h in RPMI medium supplemented with 5% heat-inactivated FBS.
Qualitative characterization of regulatory T cellsIn order to determine the immunosuppressive activity of Tregs, the IFNγ suppression assay was performed. The quality of stimulated Tregs was checked after 48 h of culture. The assay started by making co-cultures of Tregs with autologous PBMCs in a 1:1 ratio. For each co-culture, 50,000 isolated Tregs, as well as PBMCs, were used. The stimulation consisted of 50 ng/ml of phorbol 12- myristate 13-acetate, 500 ng/ml of ionomycin, and 2 ml/ml of GolgiStop (BDBiosciences, USA). After 5 h, the cells were stained to CD3 + CD4 + IFN-c + phenotype and analyzed by flow cytometry.
Chemotaxis assayTreg-cell trafficking was assessed by using a trans-well assay to measure the specific chemotactic activity. A total of 106 freshly isolated PBMCs were suspended in 200 µL RPMI and transferred into the upper chambers of 6.5-mm diameter, 5.0-µm pore-size polycarbonate membrane filter trans-well plates (Costar Corning, Cambridge, MA). RPMI (600 µL) or RPMI + CCL5 (15 ng/ml, Biologend) were added to the lower chamber. After 3 h at 37 °C, migrated cells were collected in the lower chamber, stained with anti-CD4 and anti-CD25, anti-CD127, anti-FOXP3, and anti-CD195 mAbs, and analyzed by flow cytometry analysis. The chemotactic index for Treg cells (CI-Treg) was defined as the ratio of percentages of Treg cells in CD4 + T cells of migrated cells and percentages of Treg cells in CD4 + T cells of original PBMCs.
Flow cytometric analysisCells were harvested and labeled by monoclonal antibodies against the following surface and intracellular markers: CD3, CD4, CD25, CD127, CD195 (CCR5), FOXP3, and IFN-γ. All samples were incubated in the dark for 20 min at room temperature, washed with 2 mL PBS, and centrifuged twice (1500 rpm, 6 min). Cells stained for intracellular markers were incubated with ready-to-use fixation buffer (BioLegend, USA) for 20 min in the dark at room temperature. Next, washed twice with 1 ml of freshly prepared permeabilization buffer (BioLegend, USA). Intracellularly stained cells were suspended in 100 µl of permeabilization buffer and labeled with antibodies in the same conditions as previously described. After incubation, labeled cells were washed twice with 1 ml of permeabilization buffer. All stained samples were resuspended in 400 µL PBS.
Expression of cell surface and intracellular markers was assessed using flow cytometry (LSRII, Becton Dickinson, USA) after gating on live cells determined by scatter characteristics. Data were analyzed by FACSDiva 6.0 Software (Becton Dickinson, USA). Figure 1 shows the gating strategy used for the analysis of regulatory T cell subsets.
Fig. 1
Gating strategy used to analyze Treg and CCR5 + Treg cells in PBMC. Identification of the total viable cell count of lymphocytes was assessed firstly based on forward (FSC) and side (SSC) light scatter parameters profiles designating size and granularity, respectively. Within a single, live, lymphocyte population, CD4 + lymphocytes were selected (Fig. 1a). This CD4 + cell population was further gated into CD4 + cells with the highest level of CD25 fluorescence defined as CD4 + CD25high cells (Fig. 1b). Events from the later gate are then transposed to the CD127 vs. FOXP3 dot plot. Cells with CD127-/low FOXP3 + profile were defined as a Treg cell population (Fig. 1c). Lastly, in the Treg population, those with FOXP3 + /CCR5 + profile were defined as a CCR5 + Treg cell population (Fig. 1d)
Plasma concentration of CCR5 ligandsPlasma was separated from the whole blood samples by centrifugation (2500 rpm/15 min) from all the patients and stored at − 20 °C. The plasma concentration of MCP-1, MIP-1B, and RANTES in the T1D patients (n = 48) and healthy controls (n = 22) were quantified using a commercial Quantikine HS ELISA test from R&D Systems (USA).
StatisticsAll statistical analyses were performed using Statistica 13.0 (StatSoft, Inc., USA) using either the nonparametric Wilcoxon test or the Mann–Whitney U test if not stated otherwise, with P-values less than 0.05 defined as significant. Figures were created with GraphPad Prism 8.
Comments (0)