Strains and growth media
The strain E. coli TOP10 (Invitrogen) was employed for cloning experiments, and the strain K. phaffii X-33 (Invitrogen) was selected as the host yeast strain for EG production (Table 2). E. coli strains were grown in low salt LB (1% tryptone, 0.5% NaCl, 0.5% yeast extract) supplemented with zeocin (25 µg/mL), kanamycin (50 µg/mL) or hygromycin B (50 µg/mL) when appropriate. The yeast strains were grown in YP (1% yeast extract, 2% peptone) supplemented with glucose (YPD) and/or xylose (YPX) at different concentrations. Zeocin (100 µg/mL), Geneticin (200 µg/mL), and hygromycin B (200 µg/mL) were added to the media used to cultivate K. phaffii when appropriate. LB or YP solidified with 2% agar was used to grow E. coli of K. phaffii on agar plates; the respective antibiotic was added as mentioned above. E. coli cultures were incubated at 37 °C and 220 rpm, and K. phaffii cultures were incubated at 30 °C and 200 rpm.
K. phaffii engineering for EG productionVector construction
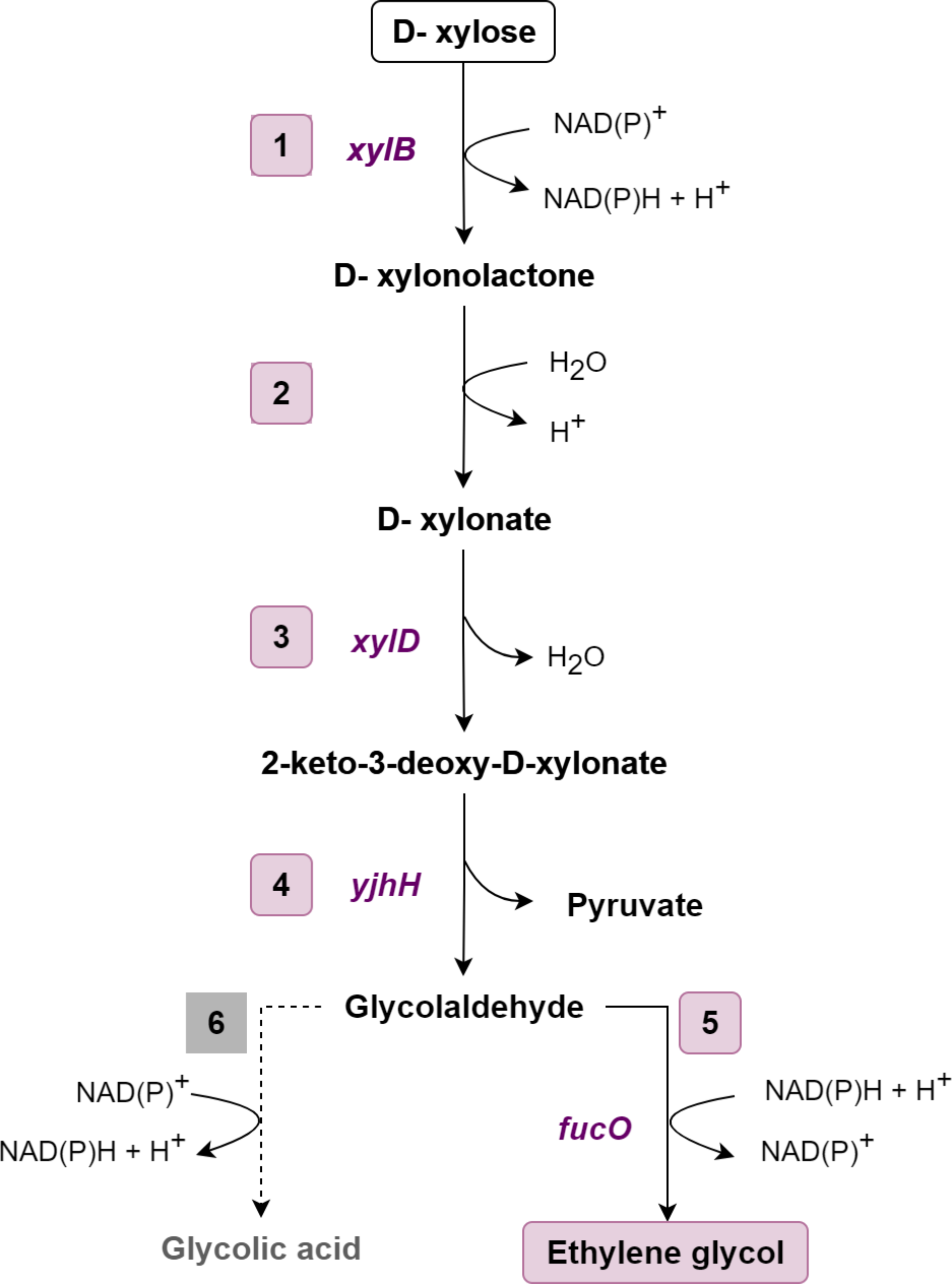
The gene xylD from Halomonas lutea (xylB-HL) encoding the first enzyme of the EG pathway (XDH) was previously cloned under the control of the PGAP promoter (pGAP-HL vector) (Ramos et al. 2021). The three gene sequences encoding putative XDs (xylD-HL, xylD-AM, and xylD-BS) were codon optimized for expression in K. phaffii, synthesized by GenOne LTDA/SA, and inserted into the pKLD vector (Betancur et al. 2017). The coding sequences (Supplementary file) were cloned under the control of the PPGK1K. phaffii promoter using the restriction sites BamHI and NotI. The two previously known XDs, encoded by yjhG-EC and xylD-CC, were used as controls in the experiments, so the same expression procedures were adopted for them. The plasmids were named pKLD-xylD-Xx, where Xx means the microorganism of origin (Table 1).
Table 1 Plasmids used in this studyThe two final enzymes of the EG-forming pathway (ALDO and ALDR) were expressed in the B3-036 vector (Prielhofer et al. 2017). For this purpose, two gene expression cassettes (named MOD1) were designed in silico and chemically synthesized (Supplementary file). For MOD1 construction, the yjhH gene encoding ALDO from E. coli was cloned under the control of the PTEF2 and CYC1 terminator using XhoI and SacI sites. The same procedure was adopted for fucO, encoding ALDR from E. coli, but the PMDH3 and TDH3 terminator regulated its expression. MOD1 was chemically synthesized, cloned and inserted into the pBSK vector by GenOne LTDA/SA. MOD1 has at the restriction site BamHI on both sides. To be expressed in K. phaffii, MOD1 was removed from the plasmid pBSK with the restriction enzyme BamHI and inserted into the B3-036 vector (Prielhofer et al. 2017), previously digested with the same enzyme, resulting in the plasmid pB3Hyg_YjhH + FucO. To obtain MOD1, the B3-036 vector was first digested with the enzymes XhoI and SacI; this reaction generated three nucleotide fragments with different sizes, 1973, 2310, and 4262 base pairs (bp). After the restriction reaction, the fragment with 4262 bp was purified with the Promega DNA purification kit. This fragment contains the cassette with the hygromycin B resistance gene (hphR) and the replication origin (PucOri). Then, the purified fragment was digested with BamHI and ligated to MOD1, resulting in the plasmid pB3Hyg_MOD1. All plasmids constructed and employed in this study are listed in Table 1.
Strain construction
The first EG pathway enzyme (XDH) was previously expressed in K. phaffii by integrating the pGAP vector carrying the xylD-HL gene (Ramos et al. 2021). For the present work, the K. phaffii strain P1HL2, already expressing XDH, was chosen to host the enzymes of the complete EG metabolic pathway. The five pKLD-xylD plasmids (Table 1) were linearized with SacI and introduced into the K. phaffii P1HL2 strain through electroporation (Invitrogen 2010). Transformants were first selected by growth on YPD plates supplemented with sorbitol (18.2%) and geneticin (200 µg/µL) at 30 °C. To further confirm the integration of the plasmid, colony PCR was carried out with the primers pPGK-D7(F) (5′-GTTCTCATCCATGAGTGAGTC-3′) and 3′AOX1(R) (5′-GCAAATGGCATTCTGACATCC-3′), resulting in amplification of the total length of the XD gene (~ 2000 bp). In addition to the 5 strains carrying the same XDH and different XDs, a control strain was also obtained by transformation with pKLD-empty (without xylD).
To construct the K. phaffii strain with the complete EG pathway, the K. phaffii strains that already had the XDH and XD integrated into the genome were transformed with the plasmid pB3Hyg_YjhH + FucO. For this, the transformation followed the same protocol described for the pKLD. After the completion of the transformation steps, the cells were spread on solid YPD plates supplemented with sorbitol (18.2%) and hygromycin B (200 µg/mL) and incubated at 30 °C until colony appearance. For confirmation, the replicative plasmid pB3Hyg_YjhH + FucO was extracted from the strains and PCR was performed with the primers PucOri(F) (5′-GATCCGGCAAACAAACCACC-3′) and CyctR (5′-CGTACACGCGTCTGTACAGA-3′). These primers amplified the first gene of MOD1 (ALDO), with a length of approximately 1900 bp. All positive clones from each construct were cryopreserved in a − 80 °C freezer. One clone of each combination of enzymes was selected for the experiments.
To construct K. phaffii strains without the heterologous fucO encoding the ALDR enzyme, digestion of the plasmid pB3Hyg_YjhH + FucO was carried out with the restriction enzyme PvuII. The enzyme cleaves the fucO protein-encoding sequence together with the MDH3 promoter and the TDH3 terminator from the plasmid pB3Hyg_YjhH + FucO. After digestion, the plasmid backbone without the fucO expression cassette was purified and joined using the T4 ligase enzyme. Once the accurate construct configuration was confirmed, the pB3Hyg_YjhH plasmid was inserted into E. coli, purified and subsequently introduced into K. phaffii through cloning. Table 2 lists all the strains selected and used in this work.
Table 2 List of strains used in this studyCultivation experiments
To evaluate the functionality of the new XD, the strains expressing XDH and one of the five XDs were incubated in YPDX medium. Initially, strains were recovered from cryogenic glycerol stocks (30%) and transferred to Petri dishes with YPD agar medium supplemented with zeocin (100 µg/mL) and geneticin (200 µg/mL). Precultures were initiated by inoculating each strain grown in solid media in 10 mL YPD medium in 50 mL conical tubes for 48 h in a shaker at 30 °C and 200 rpm. After 48 h, the preculture was transferred to 100 mL of YPD medium in 500 mL flasks for overnight growth under the same conditions of precultures. Then, cultivations were initiated by inoculating each strain to an optical density (OD600nm) equal to 10 in 30 mL YP supplemented with glucose (2 g/L) and xylose (40 g/L) in 250 mL flasks. Samples were collected at 0, 24, and 48 h of cultivation and analyzed for OD600nm, substrate consumption, and metabolite production by high-performance liquid chromatography (HPLC) and liquid chromatography coupled with mass spectrometry (LC-MS).
To evaluate the capacity of engineered K. phaffii strains to produce EG under different experimental conditions, these were cultivated as previously mentioned, except for the initial OD600nm, glucose and xylose concentrations, which varied in some experiments. The values employed are specified where appropriate. Cultivations with EG and GA as carbon sources were performed for 72 h in YP supplemented with 5 g/L EG or AG and antibiotics. Samples for OD600nm analysis, substrate consumption, and metabolite production were collected and evaluated as in previous cultivations.
Analytical methods
Quantification of glucose, EG, and glycolic acid was performed as described by Vieira (2018) and Costa et al. (2019). In brief, samples were injected in an Ultra-High-Performance Liquid Chromatography (UHPLC) system (Waters AcQuity UPLC H-Class RID) equipped with an Aminex HPX87H (Bio-Rad) column. The flow rate was adjusted to 0.6 mL/min with a mobile phase of 5 mM H2SO4. The injection sample volume was 10 µL, and the column was kept at 45 °C. The analytical run was carried out for 24 min.
Quantification of xylose, xylonic acid, xylitol, and arabitol was achieved by coupling liquid chromatography (VanquishTM, Thermo FisherTM, USA) with a mass spectrometer (Q ExactiveTM Focus orbitrap, Thermo FisherTM, USA). The samples were prepared by centrifugation at 13.800×g for 10 min. Then, the supernatant was individually filtered with a 0.2 μm filter. Subsequently, samples were injected, and metabolites were separated using a Rezex™ ROA-Organic Acid H + column (300 × 7.8 mm, 8%, Phenomenex) protected by a SecurityGuard™ Carbo H+ precolumn (4 × 3 mm, Phenomenex). The analytes were eluted with 0.1% formic acid at a constant flow rate of 0.4 mL/min and a column temperature of 80 °C. Monoisotopic mass and retention time of individually injected standards were used for analyte identification and quantification.
The qualitative analysis of KDX was performed in a Nexera X2 UHPLC system (Shimadzu, Kyoto, Japan) equipped with an Acquity UPLC HSS T3 column (2.1 mm × 100 mm, 1.8 μm) (Waters Technologies, Milford, USA) coupled to a high-resolution MaXis 4G™ Q-TOF MS analyzer (Bruker Daltonics, Germany) through an electrospray ionization source. The chromatographic run parameters were isocratic from 0.0 to 1.0 min (0% B), linear gradient from 1.0 to 3.0 min (0–5% B), linear gradient from 3.0 to 10.0 min (5–50% B), linear gradient from 10.0 to 13.0 min (50–100% B), isocratic from 13.0 to 15.0 min (100% B) and isocratic from 15.0 to 20.0 min (0% B). Eluent A was 0.1% v/v formic acid in water, and eluent B was 0.1% v/v formic acid in methanol. The flow rate, oven temperature and injection volume were 0.4 mL/min, 40 °C, and 1 µL of sample, respectively. Fermentation samples were diluted 30x before injection. Standards were injected at 30 µg/mL.
Data were acquired in negative ion mode. The MS parameters were end plate offset = 500 V, capillary voltage = 4000 V, nebulizer pressure = 4.0 Bar, dry gas flow = 9.0 L/min, dry temperature = 200 °C, spectra rate = 3.00 Hz, and detection range = m/z 75–1000. Sodium formate (2 mM) in isopropanol: water (1:1 v/v) was directly injected through a 6-port valve at the beginning of each chromatographic run for external calibration. The MS/MS parameters were collision energy, 20–50 V using a basic stepping program; cycle time of precursor ion acquisition, 3.0 s; mass range, 75–1000 m/z; spectra rate, 3 Hz; pre-pulse storage, 7.0 µs; funnel 1 RF, 300.0 Vpp. UHPLC‒MS/MS data were acquired with otofControl (Bruker Daltonics) and HyStar version 3.2 (Shimadzu) and visualized with DataAnalysis 4.2 (Bruker Daltonics). The ion extraction tool was used to manually search the m/z values of different ionization forms of some expected fermentation products in the chromatograms.
Cell growth was monitored by absorbance at OD600 with a spectrophotometer (SpectraMax M2, Molecular Devices). 5 mL aliquots were withdrawn from the starting and the last fermentation point for the cell-dry weight measurements. The aliquots were centrifuged at 3000×g for 25 min. The supernatant was discarded, and the pellet was washed in deionized water and centrifuged again. The supernatant was discarded, and the pellet was dried in a glass tube at 60 °C (approximately 96 h). Finally, the cell mass was measured with an analytical balance.


Comments (0)