
Remember me



Standard strains of C. krusei (ATCC 6258), C. parapsilosis (ATCC 22019), and the M1 protein were provided by the Infectious Disease Institute, China CDC, Beijing, China. Plasma for simulated sample preparation was obtained from Hebei General Hospital. Whole blood from healthy volunteers was collected with informed consent, anticoagulated with heparin, and centrifuged to isolate plasma, which was stored at − 80 °C. The genomic DNA was extracted from the standard strains of C. krusei and C. parapsilosis using the FastPure® Microbiome DNA Isolation Kit (Vazyme, Nanjing Vazyme Biotech Co., Ltd., Nanjing, Jiangsu, China) and stored at − 80 °C. A total of 55 blood clinical specimens from Hebei General Hospital were used to validate the performance of the DO-RAP. The specimens included 10 C. krusei-positive blood culture specimens, 15 C. parapsilosis-positive blood culture specimens, and 30 blood culture-negative specimens.
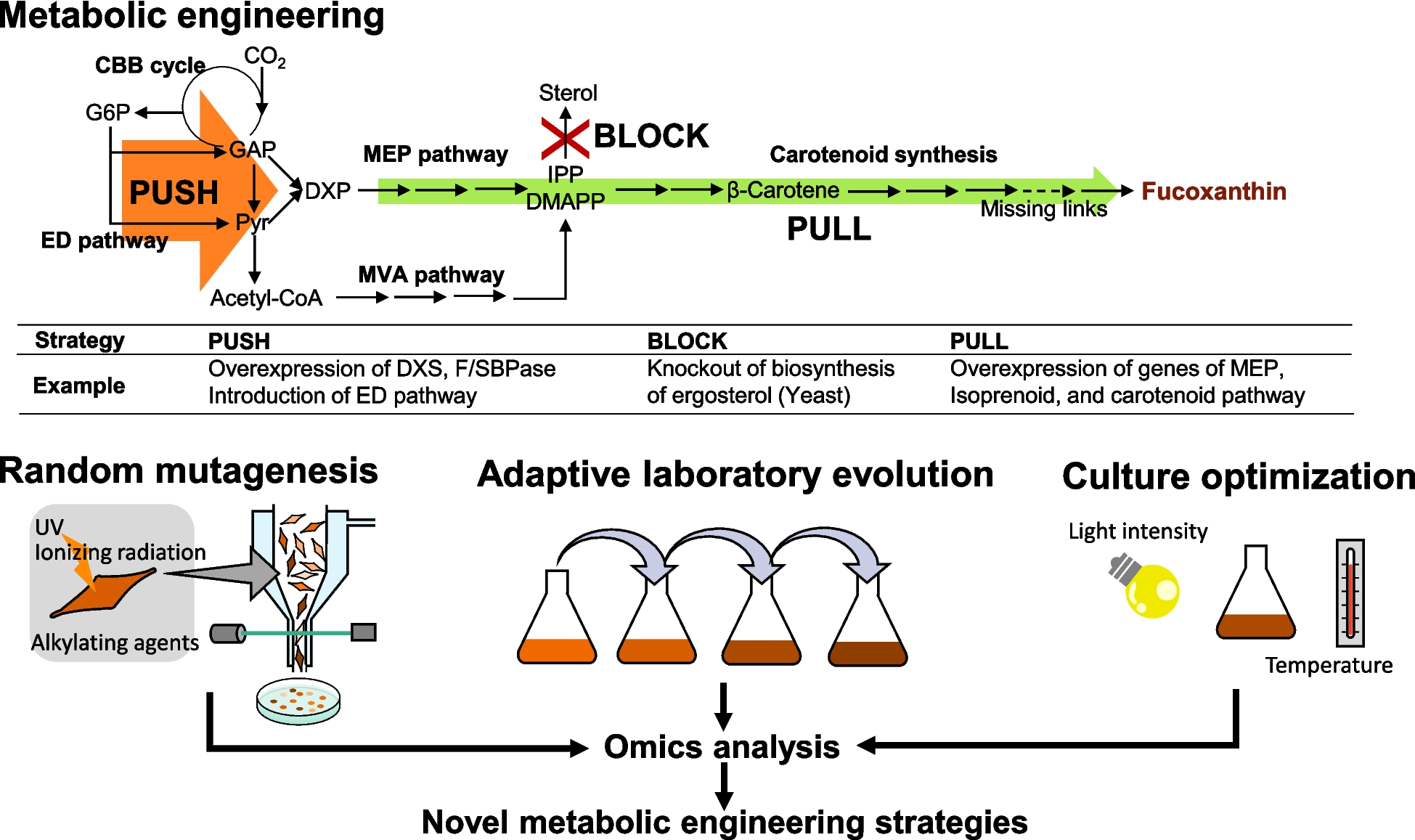
Design of DO-RAP primers, probes, and plasmid constructionPrimers and probes targeting the 26S ribosomal RNA gene in C. krusei (U767347.1) and the NADH5 mitochondrial gene in C. parapsilosis (NC_005253.2) were designed based on published literature (Brinkman et al. 2003; Vahidnia et al. 2015), in which the species-specific detection assays for nearly all known ascomycetous yeast species were developed (Brinkman et al. 2003). The RAA primers for the DO-RAP assay were designed to be 30–35 base pair (bp) in length to optimize nucleic acid-protein complex formation between the recombinase and primers, avoiding shorter primers that could hinder this process or longer primers that may promote stable primer-dimer structures. The probe for the DO-RAP assay was identical to that used in the qPCR assay. Primer design was analyzed and optimized using AmplifX (v1.7.0, CNRS, Aix-Marseille University, France. Available at: https://inp.univ-amu.fr/en/amplifx) and Oligo7 (v7.56, Molecular Biology Insights, Inc., Cascade, CO, USA) software, considering factors such as GC content, primer dimers, hairpin structures, and tertiary formations. The primers and probes were analyzed in silico to ensure specificity and to confirm the absence of cross-reactivity. Full-length sequences of C. krusei and C. parapsilosis were retrieved from NCBI, and alignments were performed using Vector NTI (v11.5.1, Invitrogen, Carlsbad, CA, USA) to identify optimal locations for the DO-RAP primers and probes. All primers and probes were synthesized by Sangon Biotech (Shanghai, China) and the sequence details are shown in Table 1. A 290 bp fragment of the conserved 26S ribosomal RNA gene from C. krusei and a 334 bp fragment of the conserved NADH5 mitochondrial gene from C. parapsilosis were inserted into the pUC57 vector (TsingKe Biotech Corp., Beijing, China) to construct recombinant plasmids. These plasmids were synthesized and provided by TsingKe Biotech Corp. (Beijing, China). The quantity of plasmid DNA was determined using the Qubit dsDNA BR/HR Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) and the Qubit 2.0 fluorometer (Life Technologies, Waltham, MA, USA). Plasmid copy numbers were calculated using the formula: DNA copy number (copies/μL) = /(DNA length × 660). The recombinant plasmids were diluted in tenfold serial concentrations ranging from 105 to 100 copies/μL in 1 × TE buffer and stored at − 80 °C until further use.
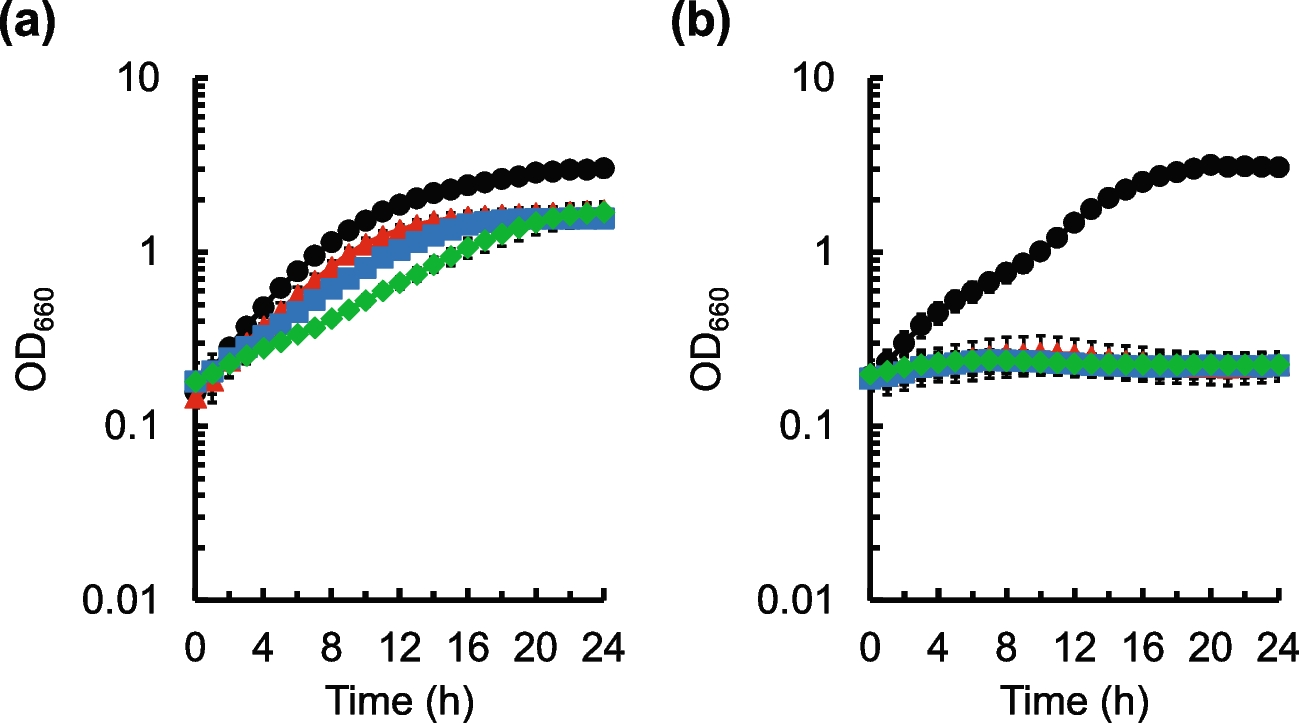
Table 1 The DO-RAP primer and probe sequencesEstablishment and optimization of the DO-RAP methodThe optimized DO-RAP reaction system consisted of 10 μL of reaction buffer and RAA enzyme mixture (Amp-Future, Amp-Future Biotech Co., Ltd., Changzhou, Jiangsu, China), 1 μL (2.5 U) of Taq DNA polymerase (Nagene Diagnosis, Beijing, China), 0.4 μL each of forward and reverse primers (10 μM), 0.2 μL each of fluorescent probes (10 μM), 0.4 μL of single-stranded DNA-binding protein (Yugong Biotech, Yugong Biolabs Co., Ltd., Jiangsu, China) (500 μg/mL), 0.4 μL of betaine (Sigma-Aldrich, St. Louis, MO, USA) (5 M), 7.2 μL of nuclease-free water, 2 μL of 100 mM magnesium ions (added to the cap of the 8-strip tube, equivalent to 8 mM magnesium), and 2 μL of recombinant plasmids or extracted genomic DNA, making a total volume up to 25 μL. The subsequent amplification process was conducted within an Archimed X6 fluorescence qPCR instrument (manufacturer: Kunpeng Gene Technology Co., Ltd., Beijing, China). The reaction program was configured as follows: an initial cycle at 40 °C for 10 min, followed by 35 cycles of 95 °C for 15 s and 60 °C for 40 s with concurrent fluorescence acquisition. The principle of the reaction is shown in Fig. 1. To optimize this DO-RAP method, we evaluated various magnesium ion concentrations, qPCR annealing duration, and RAA reaction duration, using a recombinant plasmid template diluted to 102 copies/μL.
Fig. 1
The detection principle of DO-RAP
Sensitivity, reproducibility, and specificity of the DO-RAP methodThe plasmids of C. krusei and C. parapsilosis were serially diluted in tenfold increments, ranging from 100 to 105 copies/μL. These diluted plasmids served as templates in the DO-RAP assay, with DEPC-treated water (DEPC, diethylpyrocarbonate, a reagent used to degrade RNases) employed as a negative control. Furthermore, genomic DNA from both C. krusei and C. parapsilosis was extracted in accordance with the guidelines provided by the FastPure® Microbiome DNA Isolation Kit (Vazyme, Nanjing Vazyme Biotech Co., Ltd., Nanjing, Jiangsu, China), the extracted DNA was subsequently diluted to concentrations ranging from 10−7 to 10−3 ng/μL and subjected to the aforementioned DO-RAP assay. To assess reproducibility, the DO-RAP sensitivity tests were repeated eight times at various intervals.
Furthermore, to evaluate the specificity of the method, DNA of various microorganisms closely associated with BSIs, including C. albicans, C. tropicalis, C. glabrata, Staphylococcus aureus, Pseudomonas aeruginosa, Klebsiella pneumoniae, E. coli, Staphylococcus epidermidis, Enterococcus faecium, Enterococcus faecalis, Enterobacter cloacae, Streptococcus pneumoniae, Mycobacterium tuberculosis, and Listeria monocytogenes, were then evaluated in parallel with the DO-RAP assay, which was designed to target C. krusei and C. parapsilosis. These strains are listed in Supplementary Table 1. This comprehensive approach enables a thorough assessment of the method’s specificity.
Sensitivity of quantitative polymerase chain reaction (qPCR)In order to assess and compare the sensitivity of the DO-RAP assay with that of standalone qPCR, we conducted qPCR analysis on recombinant plasmids derived from C. krusei and C. parapsilosis. The qPCR procedure was executed in strict adherence to the manufacturer’s instructions for the qPCR probe set (Entrans qPCR Probe Set V2, ABclonal, Wuhan, China). The amplification protocol encompassed an initial denaturation step at 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s and 60 °C for 1 min.
Preparation of simulated samples and M1 magnetic beadsThe standard strains of C. krusei (ATCC 6258) and C. parapsilosis (ATCC22019) were cultivated on Sabouraud agar plates (Hopebio, Qingdao Hopebio Biotechnology Co., Ltd., Qingdao, Shandong, China) and subsequently incubated in a CO2 incubator (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C for a period of 48 h. A single colony was isolated and propagated in yeast extract peptone dextrose medium (Hopebio, Qingdao Hopebio Biotechnology Co., Ltd., Qingdao, Shandong, China) by overnight shaking at 220 rpm in a shaking incubator (THZ-032, Shanghai Boqi Biological Technology Co., Ltd., Shanghai, China) at 37 °C. Subsequently, 1 mL of the culture was subjected to centrifugation, the resultant pellet was resuspended in 1 mL of phosphate-buffered saline (PBS) (GIBCO, Thermo Fisher Scientific, Waltham, MA, USA), and the pellet was then discarded. This PBS washing procedure was repeated twice. The concentration of the bacterial suspension was determined using a hemocytometer.
Simulated plasma samples were generated by introducing < 2, 4, 6, 8, 10, 50, 100, 300, 500, 700, and 900 colony-forming units (CFU) of the bacterial suspension into 1 mL of sterilized human plasma (final concentrations: < 2, 4, 6, 8, 10, 50, 100, 300, 500, 700, and 900 CFU/mL). In a similar manner, simulated PBS samples were prepared by mixing the bacterial suspension with 1 mL of PBS. These samples were then utilized to enumerate colonies on Sabouraud agar plates.
The preparation of M1 magnetic beads entailed the conjugation of 1 mg of protein A magnetic beads (Biocanal, GenScript Biotech Corporation, Nanjing, Jiangsu, China) with 1 mL of PBS. Protein A, a bacterial cell wall protein derived from S. aureus, specifically binds to the Fc region of immunoglobulin G (IgG), thereby enabling the immobilization of M1 protein on the magnetic beads. The mixture was then subjected to vortexing and subsequently separated on a magnetic stand for a period of 3 min. This was followed by the removal of the upper layer of the mixture. This process was repeated twice. The beads were then detached from the magnetic stand and resuspended in 1 mL of PBS. Subsequently, 232 µg of M1 protein was added, and the mixture was agitated for 30 min using a suspension mixer. Finally, the mixture was separated on a magnetic stand for 3 min, the upper layer was discarded, and the beads were resuspended in 100 µL of PBS. The prepared M1 magnetic beads can be stored at 4 °C for a maximum of two weeks.
Capture of Candida and genomic DNA extractionSimulated plasma samples containing 2, 4, 6, 8, and 10 CFU were aliquoted into 14-mL round-bottomed tubes, each supplemented with 1 mg of M1 magnetic beads. The volume was adjusted to 10 mL with PBS, followed by the addition of CaCl₂ (Yuanye, Shanghai Yuanye Biotechnology Co., Ltd., Shanghai, China) to a final concentration of 4 mMol/L. After thorough mixing for 1 h, magnetic separation was performed using a magnetic stand. The supernatant was discarded, and the system was washed three times with PBS to remove unbound plasma components and pathogens. The sample was then resuspended in 400 µL of PBS. Similarly, simulated PBS samples were enriched with 2, 4, 6, 8, and 10 CFU using the same method, and colony counts were obtained after overnight incubation on plates. DNA extraction from both unenriched simulated plasma samples and M1-enriched samples was performed using a DNA extraction kit (Vazyme Biotech, Nanjing Vazyme Biotech Co., Ltd., Nanjing, China). The extracted DNA was used as a template for DO-RAP and qPCR.
Evaluation of DO-RAP combined with M1 magnetic bead enrichment using simulated samplesIn this experiment, genomic DNA was extracted from varying concentrations of M1-enriched and non-enriched simulated plasma samples of C. krusei and C. parapsilosis. Extracted DNA was then analyzed using DO-RAP and qPCR. This experiment was replicated three times to ensure the reliability of the results.
Evaluation and comparison of DO-RAP and qPCR assays for clinical specimenA total of 25 clinical specimens with positive blood cultures were collected from Hebei General Hospital, including 10 specimens of C. krusei and 15 specimens of C. parapsilosis. Additionally, 30 control specimens with negative blood cultures were included. Following the extraction of genomic DNA, both the DO-RAP assay and qPCR were performed on the specimens. The consistency between the two detection methods was then compared to assess the clinical performance of the DO-RAP assay.
Statistical analysisStatistical analysis was performed using IBM SPSS Statistics (v25.0, IBM Corp., Armonk, NY, USA). Kappa analysis was utilized to evaluate the consistency between the two methods, with statistical significance set at P < 0.05.
Comments (0)