
Remember me



The hypoxia genes downloaded from MSigDB were matched to differential expression genes (DEGs) (|Log2 FC|> 1, p < 0.05) in HCC identified by analyzing mRNA expression profiles in cancer and adjacent noncancerous tissues from the TCGA database. A total of 68 DEHRGs were observed, including 46 up-regulated genes (log2 FC > 1) and 22 down-regulated genes (log2 FC < − 1) (Fig. 1A). Similarly, 434 DEIRGs were identified with the data from ImmPort (Fig. 1B), which concluded 293 up-regulated genes (log FC > 1) and 141 down-regulated genes (log FC < − 1). Among them, ten DEGs showed association with hypoxia and immune simultaneously (Fig. 1C). Then, Gene Ontology (GO) analysis, including biological processes (BP), molecular functions (MF), and cellular component (CC), was performed to investigate the potential functions and locations of DEIRGs in HCC respectively. According to BP, the up-regulated DEIRGs were supposed to participate in messenger signaling transmission, cell migration, and proliferation. However, the down-regulated DEIRGs were associated with cytokine production, cell activation, and immune response. All the DEIRGs were enriched in regulating receptor and ligand activation based on MF. As shown in CC, the DEIRGs play a part in the composition of the plasma membrane and cellular lumen, and the down-regulated DEIRGs are primarily involved in the immunoglobulin complex. According to KEGG analysis, the up-regulated DEIRGs showed strong enrichment in the Ras signaling pathway, Rap1 signaling pathway, and MAPK signaling pathway, which were involved in tumor progression. Also, the down-regulated DEIRGs were mainly enriched in protein interaction (Fig. 1D). Likewise, the up-regulated DRHRGs were mainly involved in carbohydrate catabolism in fields of biologic processes, focused on monosaccharide and cadherin binding in molecular functions, and participated in cellular composition, including the anchoring component of membranes and endoplasmic reticulum lumen. Based on KEGG analysis, they preferred to enrich in glycolysis, carbon metabolism, and HIF-1 signaling pathway. The down-regulated DRHRGs showed enrichment in carbohydrate biosynthetic process, associated with binding to R-SMAD, RNA polymerase activating transcription factor in molecular functions, and engaged in cellular component of endoplasmic reticulum lumen. Shown in KEGG result, they enriched in glycolysis and HIF-1 signaling pathway (Fig. 1E).
Fig. 1
Identification and annotation of DEGs in HCC. Volcano plot of DEHRGs (A) and DEIRGs (B) in HCC based on data from TCGA. C Venn diagram of the common genes of DEHRGs and DEIRGs. D&E. The KEGG pathways analysis and GO enrichment analysis based on biologic process, molecular function, and cellular component of DEHRGs (D) and DEIRGs (E)
Prognostic Modeling of Hypoxia-Immune-Related DEGs in the TCGA CohortPrevious studies demonstrated that hypoxia TME facilitated tumor progression [10], it is reasonable to suggest that a comprehensive analysis of hypoxia-immune-related DEGs would benefit for predicting HCC prognosis. The co-expression analysis identified 51 HDEGs and 195 IDEGs in TCGA (Fig. 2A). Among them, 78 hypoxia-immune-related DEGs (HIRDEGs) were associated with the prognosis of HCC (Table 1). To simplify the clinical examination process and remove prognostic indicators emerging from over-fitting, LASSO penalized Cox regression was used to construct the prognostic model, and a total of seven genes were determined to form a risk score conclusively (Fig. 2B and C). Among them were three immune-related genes (NR0B1, SPP1, and ADM) and four hypoxia genes (IGFBP3, SLC2A1, SERPINE1, and DPYSL4). The Kaplan–Meier (KM) analysis was performed to illuminate the predictive value of individual genes (Figure S1A–G). Further, the Spearman correlation test result indicated that almost all the seven genes were positively related and interacted with other genes excluding DPYSL4 (Figure S1H–I). The risk score = IGFBP3*0.04218 + DPYSL4*0.00067 + SLC2A1*0.07212 + SERPINE1*0.00857 + SPP1*0.01560 + NR0B1*0.04198 + ADM*0.00583. Then, we calculated the risk scores of HCC patients from TCGA; the median risk score (1.469585) was used to classify the risk subgroups. The data presented that 210 HCC patients with a risk score above 1.469585 were labeled as a high-risk group, while the remaining 210 HCC patients with risk scores below 1.469585 were recognized as a low-risk group. The result of Kaplan–Meier survival curve analysis demonstrated that the prognostic signatures of the high-risk group were significantly worse than those of the low-risk group (p < 0.001), including overall survival (OS), disease-specific survival (DSS), disease-free survival (DFS) and progression-free survival (PFS)(Fig. 2D and F–H). Further, the time-dependent area under the ROC curve (AUC) for OS at 1-, 2-, 3-, 4- and 5-years was 0.735, 0.667, 0.699, 0.703 and 0.660, respectively, which suggested that the prognostic model has favorable specificity and sensitivity (Fig. 2E).
Fig. 2
Construction of a prognostic modeling of HIRDEGs in HCC in the TCGA cohort. A Circos plots of the annotation and interaction of DEHRGs and DEIRGs in the genome. The outer circle represented the relative expression level of individual identified gene in the TCGA cohort. The center lines indicated the potential interaction predicted by the STRING database between one hypoxia gene and one immune gene, the orange color represented positive correlation and the green color represented negative correlation. B, C The LASSO Cox regression model was constructed from 78 signature genes, and the tuning parameter (λ) was calculated based on the partial likelihood deviance with tenfold cross validation. An optimal log λ value was defined at − 3.37, as shown by the vertical black lines in the plots. The seven signature genes were identified according to the best fit profile. D–H Construction of TCGA training set. D OS of TCGA HCC cohort. E Time-dependent ROC curves validation of prognostic value of the prognostic index in TCGA cohort. F–H The Kaplan–Meier survival curve of DSS, DFS, and PFS. DFS disease-free survival, DSS disease-specific survival, PFS progression-free survival, ROC receiver operating characteristic, TCGA The Cancer Genome Atlas
Table 1 Information of 78 HIRDEGs identified in this studyExternal Validation of Seven-Genes Prognostic ModelsValidation of external independent databases is essential to demonstrate prognostic models' applicability. We calculated the risk score of every patient from independent cohorts (GSE14520-GPL3921, n = 222) and classified them into high-risk and low-risk subgroups based on the cut-off value (1.469585) determined above in the TCGA cohort. The clinical data from the GSE14520-GPL3921 cohort were utilized to verify the accuracy of the prognostic model; as shown below, the OS and RFS of the two groups were different, and the high-risk group suffered worse survival status (Fig. 3A). Also, the time-dependent area under the ROC curve (AUC) for OS at 1-, 2-year in GSE1450-GPL3921 was 0.682 and 0.706, respectively (Fig. 3B), which strongly indicated the promising clinical utility of the prediction model in this study. In addition, a higher risk of death for patients with increasing risk scores was observed in both TCGA and GSE14520-GPL3921 cohorts (Fig. 3C, D). Compared to the low-risk group, the expression of the seven genes was higher in both TCGA and GSE14520-GPL3921 cohorts, excluding DPYSL4 (Fig. 3E, F).
Fig. 3
External validation of the prognostic model in predicting OS of HCC based on GSE14520-GPL3921cohort (n = 222). A, B The Kaplan–Meier survival analysis and time-dependent ROC analysis of the prognostic model for predicting the OS of HCC patients in the GSE14520-GPL3921 cohort (n = 222). C, D The distribution of the risk score, the heatmap and the survival status of patients on the TCGA cohort (C) and the GSE14520-GPL3921 cohort (n = 222) (D). E, F The box plots of the expression of the signatures in the TCGA cohort (E) and the GSE14520-GPL3921 cohort (F)
Independent Validation of the Prognostic ModelAs shown below, the risk score, T stage, M stage, and TNM stage were associated with prognosis as the result of univariate Cox regression analysis (Fig. 4A). However, multivariate Cox regression analysis only considered the risk score as an independent feature for prognosis (Fig. 4B). The data in other independent cohorts (GSE14520-GPL3921, n = 222) showed consistent with the above results (Fig. 4C and D).
Fig. 4
Independence validation of the prognostic model. A, B The forrest plot of the univariate- and the multivariate Cox analysis on the TCGA cohort. C, D The forrest plot of the univariate- and the multivariate Cox analysis on the GSE14520-GPL3921 cohort (n = 222)
Immune Profiles Between the Low-Risk and High-Risk Groups of HCC PatientsRecently, immunotherapy has attracted much attention as a novel treatment for HCC. It is reported that immunotherapeutic efficacy depends heavily on identifying the tumor microenvironment. Here, to investigate the ability of prognostic models to assess the tumor immune microenvironment of HCC, we compared the differences between the high-risk and low-risk groups of HCC patients in terms of infiltration of 22 immune cell types with CIBERSORT (Fig. 5A and B). Patients at high hypoxia-immune risk had higher proportions of M0 macrophages [11, 12], M1 macrophages [13], and monocytes [14] (Fig. 5C), of which M0 macrophages and monocytes were associated with poor prognosis of HCC (Figure S4). However, anti-tumor members were enriched in HCC patients in the low-risk group, including γδ T cells, helper T cells, and resting mast cells (Fig. 6C). Also, the expression level of the seven genes showed an association with the type of immune cells in the HCC microenvironment (Figure S2).
Fig. 5
The profiles of immune infiltration in HCC patients with different risk scores. A A heatmap showing the differences of 22 types of immune cells between high- and low- risk group. B The correlation between each type of immune cell in HCC samples was calculated with the Pearson’s correlation analysis. The size of the circle area and the gradient of colors indicated the correlation coefficient R. C The vioplot of the difference of the immune cell infiltration between high-risk and low-risk groups evaluated by the Cibersort algorithm, *p < 0.05 was considered as a standard for screening the samples. *p < 0.05, **p < 0.01, ***p < 0.001
Fig. 6
Evaluation of the cancer-immune cycle characterization genes between the two risk score groups. A, B The heatmap (A) and the box polt (B) showed the differential expression of the cancer-immune cycle characterization genes between high-risk and low-risk groups. C–E Correlation analyses between risk scores and the expression of three significantly up-regulated expressed cancer-immune cycle characterization genes, CXCL18 (C), HAVCR1 (D), VTCN1 (E). F–H Correlation analyses between risk scores and the expression of three significantly up-regulated expressed immune checkpoints, PD-1 (F), PD-L1 (G), CTLA4 (H). *p < 0.05, **p < 0.01, ***p < 0.001
Immunosuppressive Tumor Microenvironment Suggested by High Hypoxia-Immune Risk ScoreTumor immunotherapy usually features the tumor immune cycle as the fundamental framework, which focuses on the T cell-mediated cellular immune process and includes seven main steps: antigen release from dead tumor cells, antigen presentation, T cell activation, T cell migration to the tumor, T cell infiltration in tumor tissue, recognition of tumor cells by T cells and tumor cells elimination. To investigate the association of HIRDEGs with the immunosuppressive tumor microenvironment, we compared the expression of signature genes negatively regulating the processes above in high- and low-risk groups. As shown below, genes characterized as a negative regulator of the tumor immune cycle are prevalently overexpressed in high-risk groups (Fig. 6A). Among them, the expression of HAVCR1, VTCN1, and CXCL8 was significantly up-regulated and positively correlated with the risk score (Fig. 6B–E). Compared to the low-risk group, the classical immune checkpoints, including Programmed cell death-1 (PD-1), Programmed cell death ligand-1(PD-L1), and Cytotoxic T-lymphocyte-associated protein-4 (CTLA4), were significantly overexpressed in the high-risk group (Fig. 6F–H). The data above suggested that patients in the high-risk group suffered from an immunosuppressive tumor microenvironment, which may lead to worse survival.
Sensitivity of Molecular Drug Therapy Suggested by Hypoxia-Immune Risk ScoreThe primary objective of data analysis is to provide new insights and recommendations for the clinical management of HCC. As a result of drug sensitivity analysis, we found that the IC50 value of Entinostat, AZD6482, AZD818, PARP-9495, IAP-7638, and Tretinoin in the high-risk group was lower than that in the low-risk group (Fig. 7A), suggesting that these six drugs were more effective for patients in the high-risk group. In addition, patients with HCC in the low-risk group showed more sensitivity to Brivanib, Bryostain1, AZD4547, Luminespib, Mirin, JAK1-8709, GSK429286A (Fig. 7B), indicating that the seven drugs above should be considered as a priority for treatment in low-risk group.
Fig. 7
Evaluation of drug sensitivity by hypoxia-immune risk score. A, B The box plots of the estimated IC50 for thirteen chemotherapeutic drugs between the two risk score groups
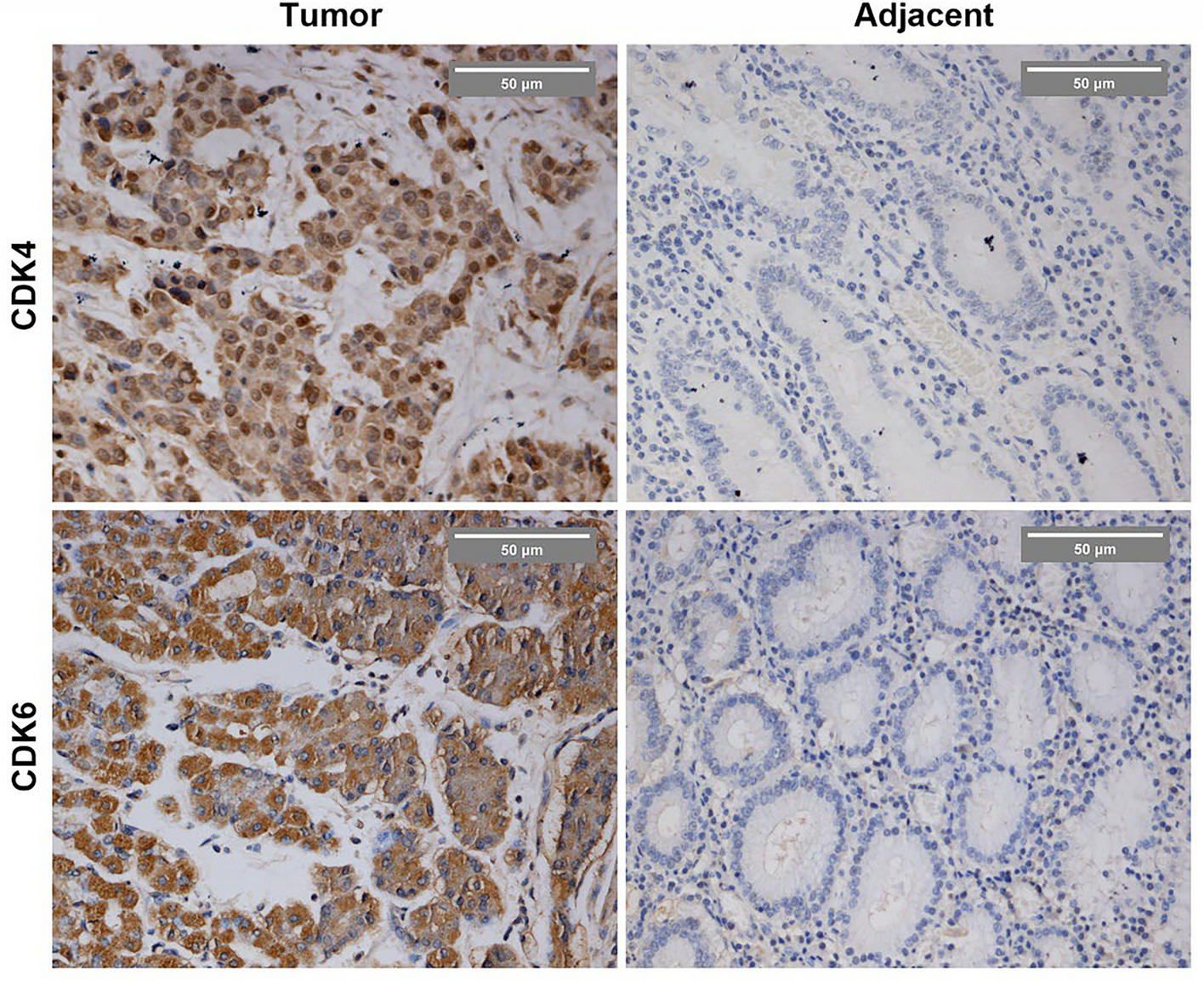
Inhibitor of Adrenomedullin (ADM)-Suppressed HCC Cell Proliferation, Migration, and InvasionTo validate the above genes in the prognostic model, we cultured HCC cells in a hypoxia environment with chloride and then examined the effect of the hypoxic environment on the expression of the above seven genes in HCC cells. The result of quantitative real-time PCR (qRT-PCR) suggested that ADM, SLC2A1, and SERPINE1 in the prognostic model were up-regulated by hypoxia, among which SLC2A1 and ADM showed the most significant (Figure S3). Since Amann et al. proved that SLC2A1 (GLUT1) was overexpressed in HCC and greatly contributed to glycolysis and cancer progression in HCC [15], we chose ADM for further studies. After comparing the expression of ADM in the normal human hepatic cell line (LO2) and several common HCC cell lines (Figure S5A), we selected HepG2 and MHCC97-H for further experiments, as they have higher ADM expression. We treated HCC cells with siRNA to knock down the expression of ADM in the MHCC97-H and HepG2 cells (Figure S5B-C) and then examined the effect of si-ADM on proliferation, migration, and invasion in HCC. As shown below, silencing the expression of ADM could suppress the ability of proliferation (Fig. 8A, B). In addition, the wounding and transwell assay results suggested a significant suppression on migratory and invasion ability of HCC in vitro (Fig. 8C, D). Instead, overexpression of ADM could promote oncogenic ability of HCC cells (Figure S6). These results strongly indicated that ADM contributed greatly to HCC development, and it should be considered a promising target for HCC treatment.
Fig. 8
Inhibition of ADM suppresses HCC cell proliferation, migration and invasion. A, B The Colony formation assay (A) and the CCK-8 analysis (B) were performed to measure the proliferation of HCC cells. C Transwell assays were performed to assess the effect of silencing ADM on the invasive and migratory ability of HCC cells. Scale bar: 100 μm. D Wound healing assays was conducted to show the effect of silencing ADM on the migratory capacity of HCC cells. Scale bar: 20 μm. (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant)
Comments (0)