
Remember me



A 52-year-old, otherwise healthy woman presented with a previously diagnosed, idiopathic, left-sided, inactive posterior uveitis. She reported occasional photopsia in the right eye, while the left eye had already developed advanced visual impairment starting at age 32, following an unclear retinitis during a one-month stay in Los Angeles. At that time, she experienced nonspecific malaise, headaches, and gastrointestinal discomfort initially deemed flu-like. She had a known history of migraine, with no evidence of mononucleosis, herpes dermatitis, weight loss, night sweats, or insect bites. She denied any recent vaccinations, tobacco use, or malignancy. Shortly before turning thirty-two, she underwent estrogen-based hormonal therapy for fertility. The patient also reported allergies to penicillin, novalgina, diclofenac, and latex.
At presentation, the best-corrected visual acuity (BCVA) measured 20/20 OD (+ 0.25/−0.25/A 11) and 20/200 OS (+ 0.25/−0.75/A 116). Intraocular pressure was 14 mmHg in both eyes, and the anterior segment showed no signs of inflammation.
Funduscopic examination of the right eye revealed a vital, sharply defined optic disc, a dry macula, and peripheral degenerations of uncertain classification. The left eye showed a slightly pale optic disc, diffuse chorioretinal atrophy with pigment shifts in the macula, and peripheral chorioretinal scars.
Optical coherence tomography (OCT) of the right eye demonstrated only a minimally blurred foveal ellipsoid zone without disruption of the outer retina (Fig. 1a, a′, “Baseline”). In the left eye (Fig. 1b, “Baseline”), OCT revealed extensive para- to perifoveal atrophy of the outer retinal layers, irregular outer layering from sub- to perifoveal, and mild macular thickening.
Fig. 1
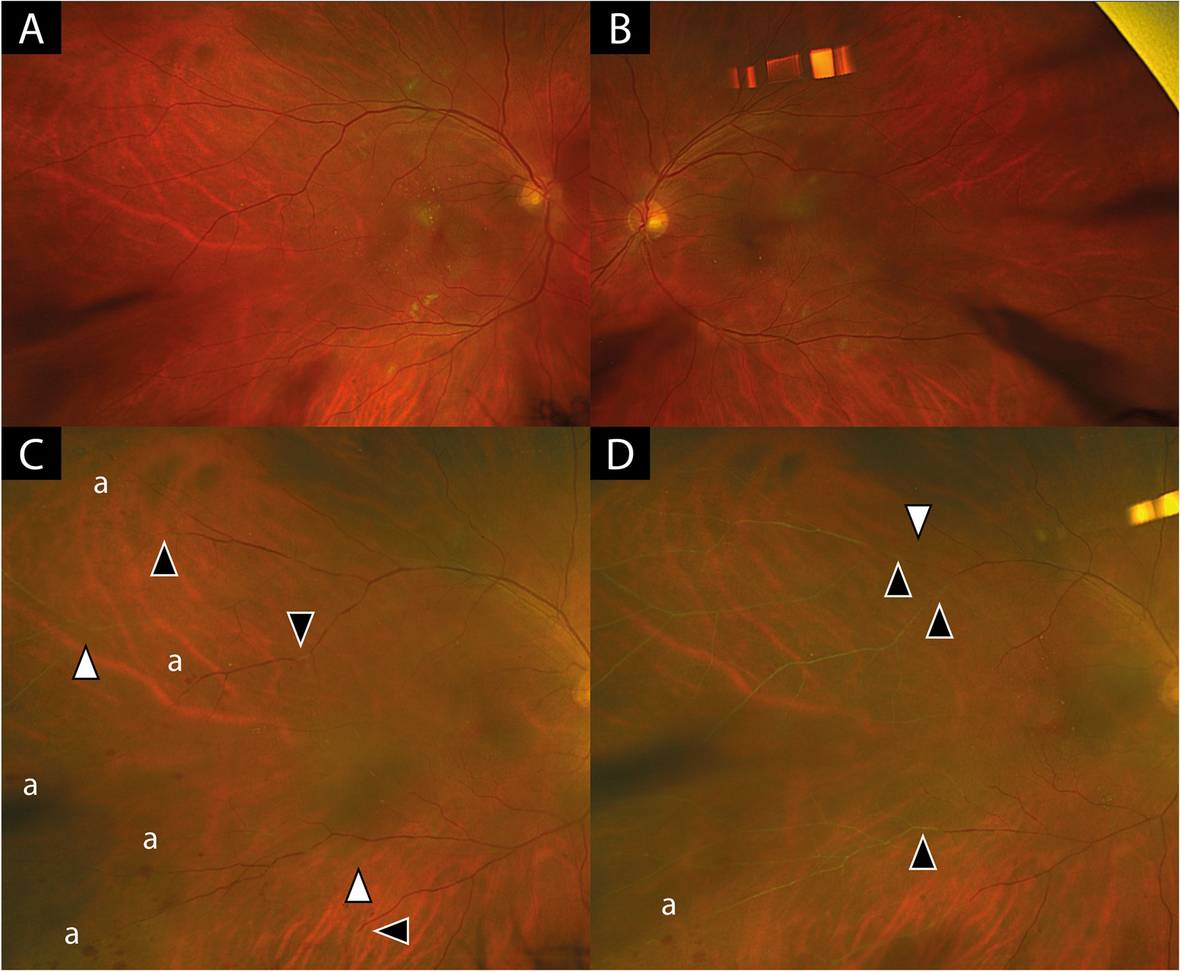
Fundus autofluorescence (FAF; Spectralis, Heidelberg Engineering, Heidelberg: a-c, a´-c, Clarus 500, Zeiss, Oberkochen, Germany). Both eyes over time (Upper row, a-d, right eye, lower row, a’-d’, left eye) The figure shows a peripapillary lesion with a trizonal autofluorescent pattern in the right eye: linear to spotty flaring hyperautofluorescent border between MORR lesion and normal retina (Zone I), speckled hyperautofluorescence within the MORR lesion (Zone II) and hypoautofluorescent area corresponding to RPE to choroidal atrophy (Zone III), with a progressive course over the years (upper row). At the 12th year of presentation, centripetal spread from inferior with mild speckled hyperfluorescent border is seen (white triangle). At the 13th year of presentation, the centripetal multizonal spread formed a subtotal circular pattern with confluent areas and showed speckled hyperautofluorescence at the margin at the transition to normal retina. The periphery showed confluent hypoautofluorescence corresponding to the extensive RPE atrophy (white triangle). The left eye shows similar absent autofluorescence over the years due to central retinal to choroidal atrophy. Individual patchy hypo- to isoautofluorescent lesions correspond to the inhomogeneously distributed pigment clumping (lower row)
Course, diagnosis and treatmentAt age 60, eight years later, the patient returned with progressive visual impairment, flickering, temporal field restriction, and photopsia in the right (better) eye. In the left eye, dark/gray vision remained stable. She reported no additional systemic infections but had HLA-B27-positive spondylarthritis and stress-related complaints. Best-corrected visual acuity dropped to 20/30 OD and light perception OS.
Funduscopic examination of the right eye (Fig. 2a-e) revealed a sharply defined optic disc, chorioatrophy from peripapillary to nasal parafoveal, dry macula with pigment shifts, and chorioatrophic scars. A distinct transition zone between atrophic and normal retina was evident (red arrowheads). By contrast, the left eye (Fig. 2g–k) exhibited a pale optic disc, diffuse atrophy, and patchy, irregular pigment clumping.
Fig. 2
Correlation of FFA, ICGA and SD-OCT for photoreceptor, RPE and choriocapillaris atrophy zones The white arrows show three border transitions in the right eye (a, c): (I) from normal anatomical outer retinal structures and choriocapillaris to (II) RPE and photoreceptor degeneration/remodeling with reticular-like drusenoid deposits in OCT and corresponding hypo- or hyperfluorescent appearance in fluorescence angiography depending on their position in relation to the pigment layer of the retina and the amount of lipids and proteins they contain, missing drusen fluorescence in RPE atrophy. Subsequently (III) complete RPE loss and limiting to absent visualization of the choriocapillaris with decrease of peripapillary choroidal thickness in OCT, corresponding window defect and simultaneously hypofluorescent in FFA, as well as peripapillary vessel density decrease (yellow arrows) of the choroid in ICG
Blue fundus autofluorescence (BAF, Spectralis®, Heidelberg Engineering, Germany) of the right eye (Fig. 3a) showed a central trizonal autofluorescent pattern: A linear to punctually flaring hyperautofluorescent border between the lesion and normal retina (Zone I), a speckled hyperautofluorescence within the lesion (Zone II), and a hypoautofluorescent area corresponding to RPE and choroidal atrophy (Zone III). The left eye (Fig. 3a’) lacked any apparent autofluorescence due to central retinal and choroidal atrophy.
Fig. 3
∆: SD-OCT progression over 13 years
On OCT, the right eye (Fig. 1c, c’, enlarged macular area) showed a nasal outer retinal thinning and ellipsoid zone (EZ) disruption. The RPE-Bruch’s membrane complex appeared fragmented, with hyperreflective spots. The external limiting membrane (ELM) can be traced to perifoveal nasal. The outer nuclear layer (ONL) appeared shortened, due to layer shifting and the partly apparent elongation of the outer plexiform layer (OPL). By contrast, the left eye (Fig. 1d) displayed complete outerretinal atrophy and markedly reduced choroidal thickness.
Fluorescein angiography (FFA) of the right eye (Fig. 4a) showed delayed vessel filling (armretina time 36 s, measured at the right eye), a peripapillary hypofluorescence and subsequent diffuse hyperfluorescence; indocyaninegreen angiography (ICGA) revealed reduced choroidal vessel density. In the left eye (Fig. 4b), severe atrophy abolished retinal fluorescence and produced marked choroidal hypofluorescence.
Fig. 4
∆: Fundus photographs demonstrating progressive atrophy and pigment changes in both eyes over time
The corresponding peripapillary OCT of the right eye (Fig. 4c) showed three transition zones: (I) From normal outer retinal structure and intact choriocapillaris, (II) to RPE and photoreceptor degeneration or remodeling with reticular-like drusenoid deposits, (III) to complete RPE loss with only limited or absent visualization of the choriocapillaris and reduced peripapillary choroidal thickness.
Peripapillary OCT (Fig. 4d) confirmed in the left eye the extensive damage to the outer retinal layers and an almost complete loss of the peripapillary choroidal layer.
Electrophysiological testing: In the right eye, multifocal ERG revealed intact central amplitudes but prolonged implicit time. By contrast, the left eye showed markedly reduced amplitudes, indicating severe global dysfunction. Full-field ERG was age-appropriate in the right eye but severely diminished in the left, with reduced amplitudes and prolonged latencies under both scotopic and photopic conditions. EOG yielded an Arden quotient of 2.2 (right) and 1.4 (left). Pattern VEP latencies were normal in both eyes; however, left-eye amplitudes were notably reduced.
With the abovementioned taken together, the findings thus pointed further towards the diagnosis of AZOOR.
Systemic evaluation ruled out significant viral/infectious causes (e.g., HIV, VZV, CMV, EBV).
Systemic comorbidities included mild HLA-B27-positive spondylarthritis, degenerative skeletal changes with right-convex scoliosis, isolated enthesopathies including diabetes mellitus and substituted hypothyroidism. Neurological co-assessment revealed no pathological findings.
To exclude paraneoplasia, comprehensive laboratory tests were performed, including hormonal assessments (FSH, LH, growth hormone, prolactin, IGF-1, and androstenedione), all of which were normal. Cancer-associated retinopathy was effectively ruled out by the absence of enolase, recoverin, and Hu-antibodies (anti-Hu/ANNA-1). MRI of the upper abdomen and pelvis showed no tumor processes. Dermatological examination revealed only seborrheic keratosis and compound nevi, with no suspicious lesions. Apart from asymptomatic reflux esophagitis, gastro- and colonoscopy were likewise unremarkable. A bone scan indicated no metastatic lesions, showing only focal activity consistent with degenerative joint and spinal changes.
Given macular involvement, systemic prednisolone (100 mg/day for 5 days, taper over 12 weeks) was administered, followed by attempts at immunomodulation (methotrexate, then adalimumab) in cooperation with the rheumatology clinic in Püttlingen, but adverse effects led to discontinuation. During immunomodulation therapy, the patient experienced severe hair loss, persistent gastrointestinal upset, headaches, pronounced fatigue with myalgia, a constant flulike malaise, and newonset depressive symptoms that led to social withdrawal.
To enhance her visual support, the patient received an electronic handheld magnifier (“pebble” from Enhanced Vision), a 7× illuminated handheld magnifier (Mobilux, Eschenbach), magnification software (ZoomText), and information on smartphone apps and other daily living aids (Marland catalog). The 3.5× Mobilux handheld magnifier with a stand proved the most comfortable solution.
Further follow-ups (+ 9, + 12, +13 years) showed continued progression on OCT (OD: Fig. 1e, e’, 1 g, g’, 1 i, i´,/OS: Figure 1f, h and j), and autofluorescence (OD: Fig. 3b-d/OS: b´- d´), predominantly in the right eye, with residual atrophy in the left. Despite therapy, vision deteriorated to light perception or worse in both eyes.
From these extensive, multizonal findings, MORR was diagnosed, representing a variant of AZOOR.
Comments (0)