Chemicals and reagents
RPMI-1640 (Gibco™, 31800-014, USA) and fetal bovine serum (FBS, Gibco™, 10437-028, USA) were procured from GIBCO-BRL. Chloroquine was obtained from Cell Signaling Technology Inc. (CST, 14774, USA), while rapamycin was purchased from Selleck Chemicals LLC (Selleckchem, S1039, USA). BPA-fructose was synthesized by Taiwan Biotech Inc. and provided by the Nuclear Science and Technology Development Center at National Tsing Hua University.
Cell culture
LUSC cell lines H2170 (ATCC, CRL-5928), H520 (ATCC, HTB-182), and the lung carcinoma cell line A549 (ATCC, CCL-185) were obtained from the American Type Culture Collection (https://www.atcc.org/) [22]. H2170-RAPA cells were derived from H2170 cells treated with the mTORC1 inhibitor rapamycin (20 μM) for 1 month, and surviving cells were pooled. All cells were cultured in RPMI-1640 medium supplemented with L-glutamine (4 mM), sodium pyruvate (1 mM), HEPES (10 mM), sodium bicarbonate (23.8 mM), penicillin/streptomycin (1%), and FBS (10%). The cells were maintained at 37 °C with 5% CO2 in a humidified atmosphere. Short tandem repeat (STR) profiling was used to authenticate all cell lines within the last three years.
Plasmids
The shRNA clone of pLKO.1-shSLC7A5 (TRC N0000043009) was obtained from the National RNAi Core Facility, Academia Sinica (Taiwan). The pLKO.1-Scrambled shRNA (#136035) was sourced from Addgene (USA).
Reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cultured cells using the TRIzol reagent (Invitrogen™, 15596026, USA) following the manufacturer’s protocol. Subsequently, cDNA synthesis was performed using 1 µg of total RNA and the PrimeScript™ RT Reagent Kit (Takara Bio USA, RR037B, USA) according to the manufacturer’s instructions. For quantitative analysis, 1000 ng of cDNA was mixed with Taqman or UPL probes and corresponding primers in each reaction. The reactions were performed in triplicate using the StepOnePlus system from Applied Biosystems. The expression levels of the genes were normalized to the housekeeping gene (18S rRNA). Relative gene expression was analyzed using the 2-ΔΔCT method. The primer and probe sequences are provided in Table S1.
Immunoblotting
Cells were collected using a lysis buffer containing a cOmplete™, EDTA-free Protease Inhibitor Cocktail (Roche, 04693132001, Switzerland). Immunoblotting was conducted using the following primary antibodies: anti-SLC7A5 (Cell Signaling, CST#5347, USA), anti-SLC3A2 (Bioss, bs-6659R, USA), anti-SQSTM1 (GeneTex Inc., GTX100685, USA), anti-LC3B (GeneTex Inc., GTX127375, USA), anti-alpha Tubulin (GeneTex Inc., GTX112141, USA), and anti-GAPDH (GeneTex Inc., GTX100118, USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were employed, and then protein bands were detected using chemiluminescence and imaging with an Image Quant LAS 4000 mini chemiluminator.
Clonogenic assay
Cells were seeded in 6-well plates at a density of 500 to 2000 cells per well, with three independent replicates for each condition. The plates were then incubated for 2 weeks at 37 °C with 5% CO2 to allow colony formation. Following incubation, the colonies were fixed and stained using a mixture of 3.7% formaldehyde, 80% methanol, and 0.25% crystal violet (Fisher Chemical, C58125, USA) for 20 min at room temperature. The area of the stained colonies was subsequently quantified using Image J software.
Cell-cycle analysis
Cells were seeded at a density of 3 × 10⁵ cells in 60 mm dishes, with three independent replicates, and incubated overnight. Subsequently, the cells were fixed by incubating them overnight with 70% ethanol at −20 °C. Following fixation, the cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) for 30 min at room temperature. The stained cells were then analyzed using flow cytometry according to the manufacturer’s instructions.
Apoptosis analysis
Cells were plated at a density of 1×105 cells in a 60-mm dish, with three independent replicates. After 48 h of incubation, the cells were harvested and fixed in 500 μL of Annexin V binding buffer, supplemented with 1 μL of Annexin V-FITC (Elabscience®, E-CK-A111, USA) and 5 μL of 7-AAD-PC5.5 (Elabscience®, CK-A162, USA). Following a 20-min incubation at room temperature, flow cytometry analysis was performed on 10,000 cells, following the manufacturer’s instructions.
Generation of SLC7A5 knockout and knockdown cell lines
The CRISPR/Cas9 system was used to generate SLC7A5 knockout H2170 cells. A guide RNA (gRNA) targeting the first exon of SLC7A5 (sequence: GGCCGGTGCGCAGAGCATGGCGG) was designed by Dharmacon Edit-R™ CRISPR-Cas9 Gene Engineering System (https://horizondiscovery.com/en). Before Cas9/gRNA delivery, H2170 cells were synchronized with 200 ng/mL nocodazole (Cayman Chemical Inc., 13857, USA) for 17 h to enhance transfection efficiency. On the day of electroporation, crRNA and tracrRNA (150 pmol each) were mixed to form the gRNA complex. Cas9 protein (30 pmol) and gRNA (150 pmol) were then combined in Opti-MEM™ I Reduced Serum Medium at room temperature for 15 min to assemble Cas9/gRNA ribonucleoprotein (RNP) complexes. Synchronized cells (1×105) were collected, washed, and resuspended in Opti-MEM (Gibco, 31985070, USA), followed by adding the RNP complexes. Electroporation was performed using the Super Electroporator NEPA21 Type II (Nepa Gene Inc., Japan) under optimized voltage conditions. Post-electroporation, cells were transferred to 24-well plates with culture medium and incubated at 37 °C with 5% CO2 for 48 h. High-resolution melting quantitative PCR (HRM-qPCR) was employed alongside the T7 Endonuclease I (T7E1) assay to assess the efficiency of CRISPR/Cas9 gene editing. The T7E1 assay detects mismatches caused by indels at the target site, visualized via gel electrophoresis. HRM-qPCR distinguishes wild-type and indel-containing DNA, enabling screening for SLC7A5 knockout cells. Gene editing was confirmed, and single-cell clones were isolated, cultured, and analyzed by HRM-qPCR and sequencing. Protein expression was assessed by immunoblotting, and positive clones were selected for further experiments. We assessed the impact of SLC7A5 knockdown on cell growth and apoptosis in pooled H2170 cells transduced with lentiviral particles encoding SLC7A5-targeting shRNA (shSLC7A5). Knockdown of SLC7A5 in pooled populations induced apoptosis, thereby limiting the feasibility of downstream analyses. To overcome this, stable SLC7A5 knockdown H2170 cells were generated through clonal selection following lentiviral transduction. A clone with confirmed SLC7A5 downregulation was selected for further experiments.
Immunohistochemistry
The immunohistochemical analysis of SLC7A5 expression in lung cancer samples was conducted using a lung cancer tissue array slide (Superbiochips, CC5, Korea). After deparaffinization, endogenous peroxidase activity was blocked by incubating the slide in methanol with 3% hydrogen peroxide (H₂O₂). Antigen retrieval was performed using sodium citrate buffer and autoclave heating. To prevent non-specific binding, the slide was incubated with 2.5% FBS. Primary antibodies against SLC7A5/LAT (Abcam, ab208776, UK) were then applied, followed by incubation with secondary antibodies. The antigen-antibody complexes were visualized using 3,3’-diaminobenzidine (DAB), and the tissue sections were counterstained with hematoxylin. Immunohistochemical staining intensities and percentages were evaluated by Dr. Shien-Tung Pan at the Department of Pathology, China Medical University Hospital. The intensity of staining was scored on a scale from 0 (no staining) to 3 (strong staining), while the percentage of stained cells was scored as 0 (<10%), 1 (11-50%), 2 (51-80%), and 4 (>80%). The tissue staining patterns were quantified using the Immunoreactive Score (IRS) assessment.
Confocal immunofluorescence microscopy
1 × 105 cells/well were seeded in 8-well chambered coverslips (Ibidi Inc., 80806, Germany) and incubated at 37 °C in a 5% CO2 incubator for 24 h. After incubation, the cells were treated with chloroquine (30 μM) for 24 h. Subsequently, the cells were stained with an anti-LC3B polyclonal antibody (1/100 dilution; GeneTex Inc., GTX127375, USA), followed by incubation with Goat anti-Rabbit IgG (H + L) secondary antibodies, DyLight™ 488 (1/200 dilution; Thermo Scientific™, 35552, USA). The nucleus was stained with Hoechst 33342 (Thermo Scientific™, 62249, USA). The slides were then analyzed under a confocal laser-scanning microscope (FluoView FV10i; Olympus, Japan), and the images were processed using FV31s-SW viewer (Version 2.4.1.198; Olympus, Japan). The number of autophagosome puncta was quantified using ImageJ software.
Boron neutron capture therapy (BNCT)
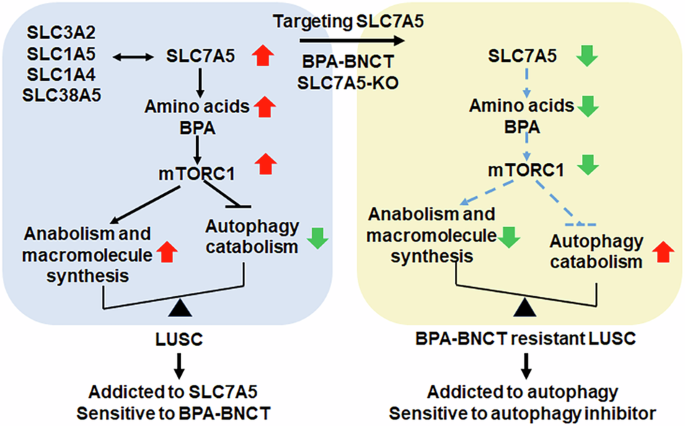
Cells were seeded at a density of 2.0 × 105 cells per well, with three independent replicates, in a 6-well polystyrene plate. After incubation for one day, cells were treated with or without a BPA-fructose solution (25 µg/mL) for 4 h, followed by three washes with cold PBS. Subsequently, the cells were irradiated with an epithermal neutron flux of 1.28 × 109nth/cm2s at an ambient temperature of 25 ± 3 °C. This irradiation was conducted at the Tsing Hua Open Pool Reactor at the Nuclear Science and Technology Development Center, National Tsing Hua University, Taiwan.
Public domain data and statistical analysis
The public gene expression profiling and RNA-seq datasets used in this study were analyzed as described [23]. The sources of these datasets are listed in Table S2. Statistical analyses were conducted using the Student’s t-test for comparing two samples or one-way analysis of variance (ANOVA) for comparing multiple samples. The overall survival of lung cancer patients was analyzed using the log-rank test. A linear regression test examined the correlation between genes in the scatter plot results. A p-value of less than 0.05 was considered statistically significant. For cell experiments, statistical analyses were carried out using Prism software (GraphPad Inc., USA). Statistical significance was defined as a p-value less than 0.05 (p < 0.05).
Ethics statement
The CC5 lung cancer tissue array used in this study was obtained from SuperBiochips and acquired in accordance with the supplier’s ethical sourcing standards. This commercially available array contains no identifiable patient information and does not require additional consent for research use. All experimental procedures, including the acquisition and analysis of publicly available datasets and commercial biospecimens, were conducted in accordance with relevant institutional, national, and international ethical guidelines and regulations.

Comments (0)