
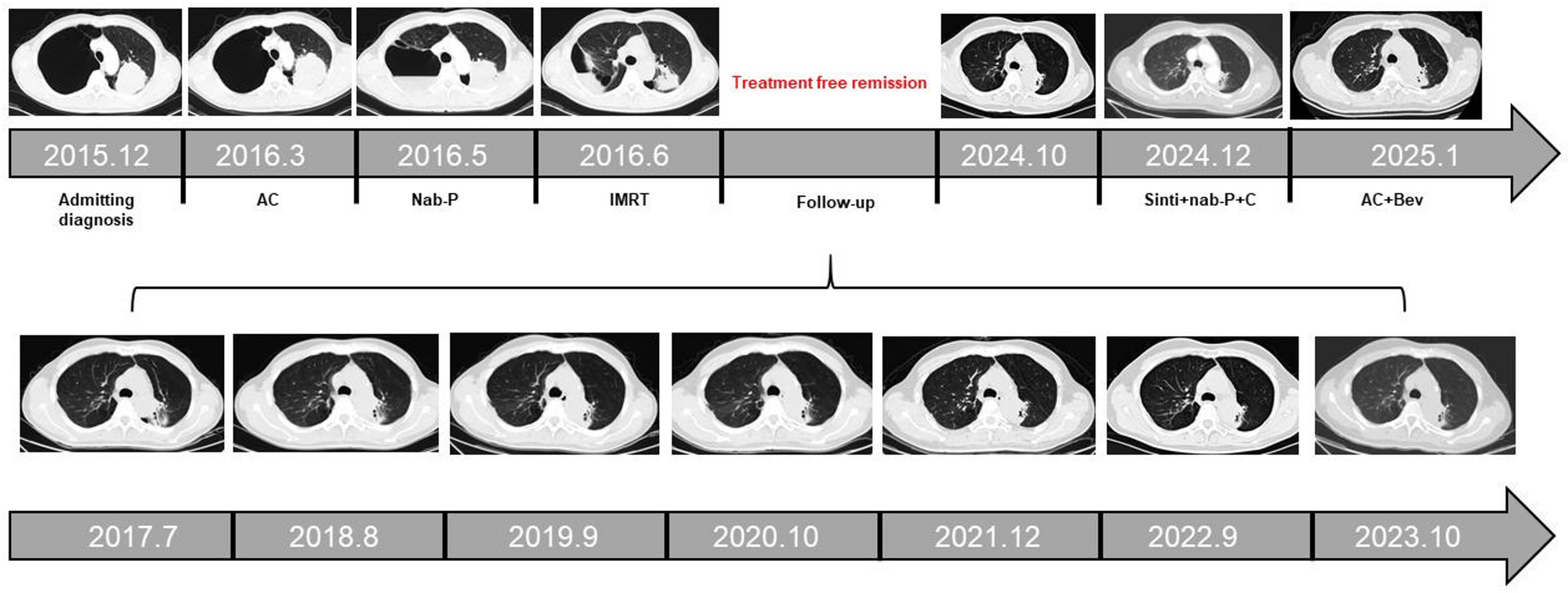
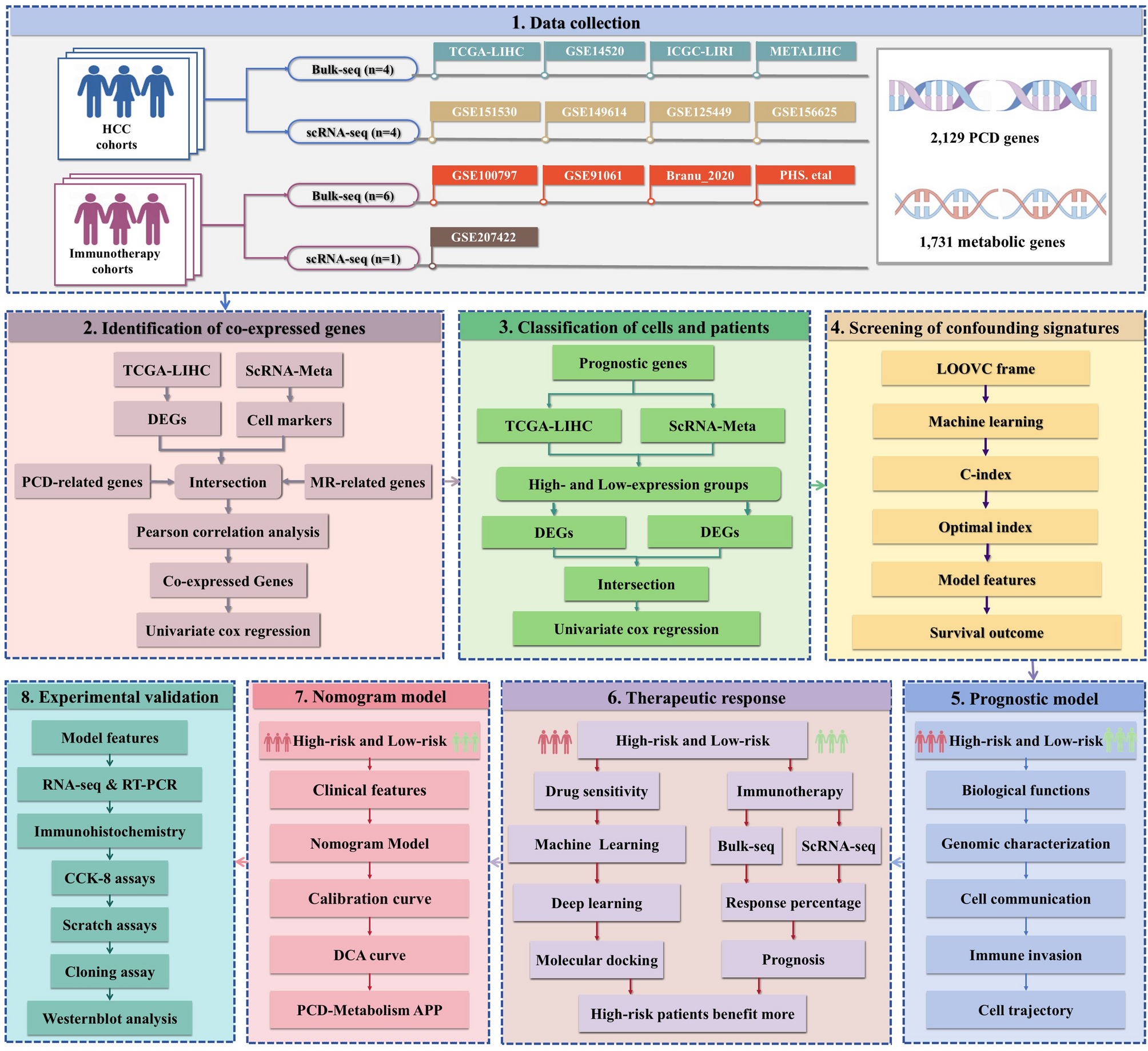
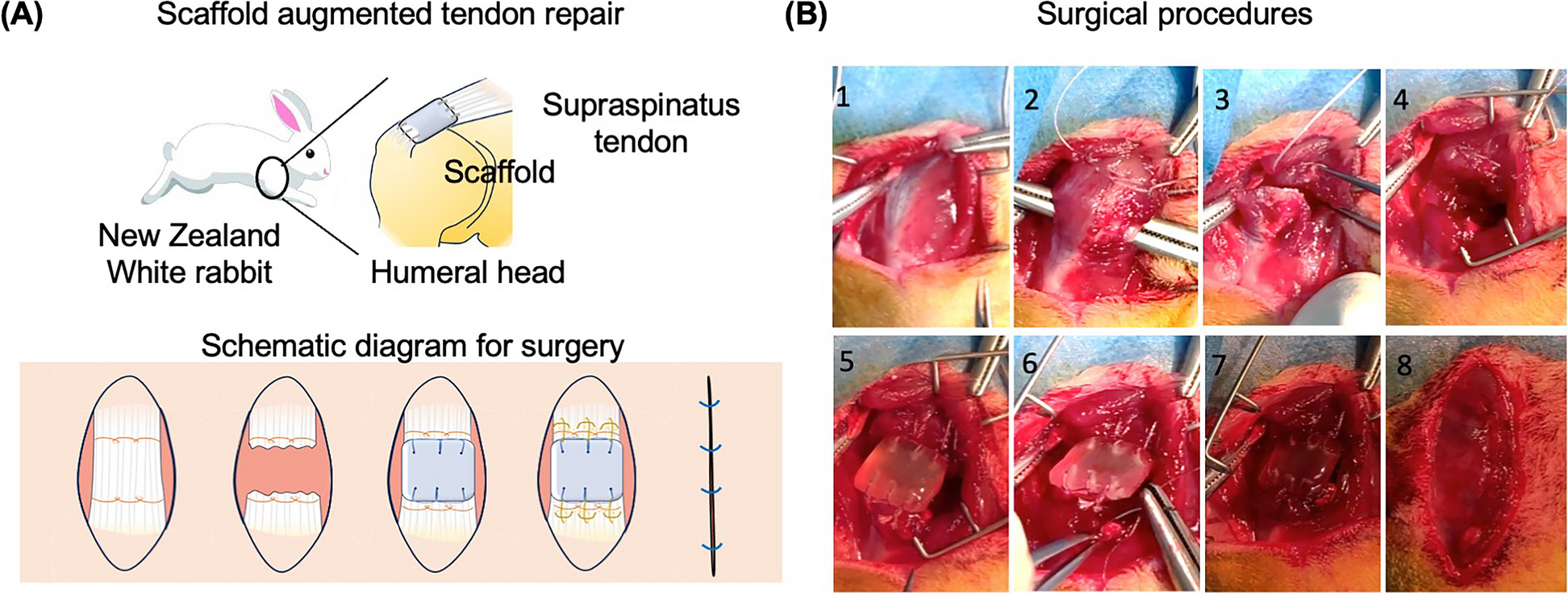
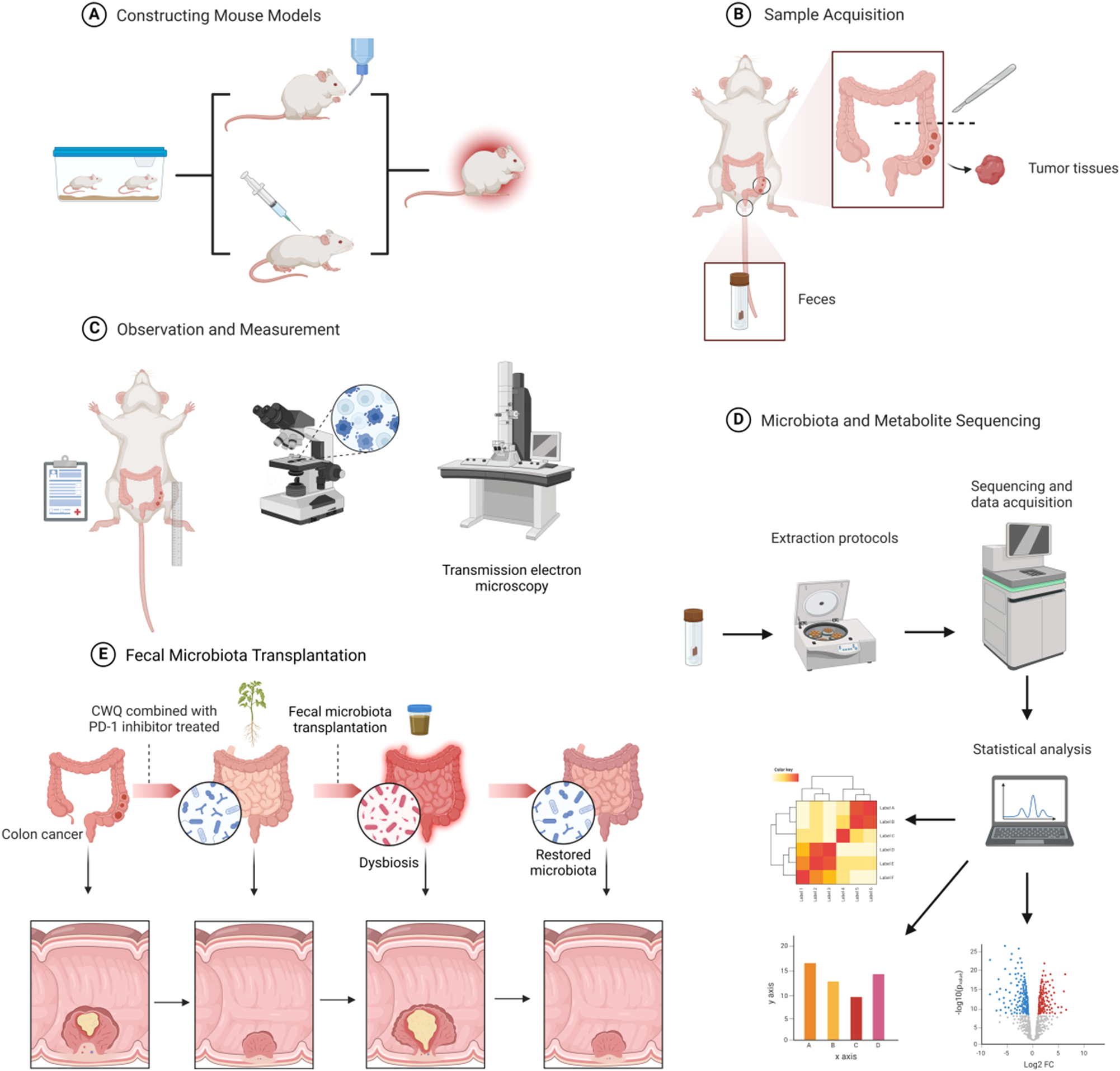
Remember me




We analyzed transcriptomic data from The Cancer Genome Atlas (TCGA) as the training dataset and the Gene Expression Omnibus (GEO) dataset GSE39582 as the validation cohort to assess the prognostic significance of peroxisomal activity in colon adenocarcinoma (COAD). A total of 106 peroxisome-related genes (PRGs) were common to both datasets and selected for downstream analysis. Univariate Cox regression analysis in the TCGA-COAD samples (n = 434) identified seven PRGs significantly associated with patient survival (Fig. 1A). Among them, ABCD1 (ATP Binding Cassette Subfamily D Member 1), FNDC5 (Fibronectin Type III Domain Containing 5), HAO2 (Hydroxyacid Oxidase 2), and PEX5L (Peroxisomal Biogenesis Factor 5 Like) were linked to a worse prognosis (risky genes), while ABCD3 (ATP Binding Cassette Subfamily D Member 3), ACOX1 (Acyl-CoA Oxidase 1), and NOS2 (Nitric Oxide Synthase 2) were associated with a favorable prognosis (protective genes). We applied LASSO (Least Absolute Shrinkage and Selection Operator) regression, a regularization technique that selects the most relevant features by shrinking the coefficients of less important variables to zero, thereby helping to identify a robust subset of prognostic genes while minimizing overfitting. (Fig. 1B-C). A risk score was calculated for each patient based on the expression levels of the seven PRGs and their respective regression coefficients obtained via lasso regression analysis. The distribution of risk scores across individuals in both the training and validation cohorts, along with their survival status (alive or deceased) and the expression patterns of the seven PRGs, is presented in Fig. 1D-E. Notably, risky genes (ABCD1, FNDC5, HAO2, and PEX5L) were upregulated in high-risk patients, whereas protective genes (ABCD3, ACOX1, and NOS2) exhibited higher expression in the low-risk subgroup. Survival analysis confirmed that patients in the high-risk group exhibited significantly worse overall survival in both datasets (Fig. 1F-G), highlighting the potential of peroxisome-related genes as prognostic biomarkers in COAD.
Fig. 1
Classification of Colorectal Cancer (CRC) Subtypes Based on Peroxisome-Related Genes (PRGs). A Forest plot displaying the results of univariate Cox regression analysis of PRGs in TCGA-COAD samples (n = 434). B-C Risk stratification of CRC patients using LASSO-penalized Cox regression based on PRG expression. D Risk score distribution, survival status, and heatmap of PRG expression in the TCGA COAD cohort (n = 434). E Corresponding risk plot and heatmap in the GEO validation cohort (GSE39582; n = 579). F-G Kaplan–Meier survival curves for high- and low-risk groups in the TCGA COAD cohort and the GEO validation dataset, respectively
Clinical Characteristics of PRG SubtypesWe then examined the clinical differences among the identified PRG subtypes and compared them with established molecular subtypes of CRC. There was no significant difference in age and gender distribution among PRG subtypes (Fig. 2A). However, a notable difference was observed in cancer stage, particularly in nodal status (N parameter). The high-risk subgroup had a greater proportion of patients with stage III/IV disease and N3 nodal involvement (Fig. 2A). The relationship between PRG subtypes and established CRC classifications was further examined, including microsatellite instability (MSI) status, Consensus Molecular Subtypes (CMS), and the CpG island methylator phenotype (CIMP). The high-risk PRG subtype was associated with a higher proportion of MSI-positive cases (Fig. 2B). In terms of CMS classification, the high-risk subgroup was predominantly CMS4, which corresponds to the mesenchymal subtype and is linked to poor prognosis, whereas the low-risk PRG subtype was mainly CMS2, corresponding to the Canonical subtype characterized by better differentiation and prognosis (Fig. 2C). The high-risk subgroup also had a higher prevalence of CIMP-High (20%), while the low-risk subgroup was predominantly CIMP-Low (32%) (Fig. 2D). These findings highlight distinct molecular and clinical characteristics of PRG subtypes, reinforcing their relevance in CRC classification and prognosis. These results highlight high-risk as a distinct subgroup characterized by strong epigenetic regulation, immune activity, and mesenchymal features.
Fig. 2
Clinical and Molecular Landscape of PRG Subtypes. A Circular plot visualizing the distribution of clinical variables—age, gender, and tumor stage—across the identified PRG subtypes, with statistical significance assessed using the chi-square test (P < 0.05). B-D Bar charts illustrating the association between PRG subtypes and major molecular classifications of colorectal cancer, including microsatellite instability (MSI), consensus molecular subtypes (CMS), and CpG island methylator phenotype (CIMP). Chi-square test (P < 0.05)
Mutational Landscape of the PRG SubtypeWe conducted an in-depth analysis of genomic aberrations across the PRG subtypes to uncover key mutational patterns. The oncoplot visualizations highlight distinct mutation distributions between PRG subtypes (Fig. 3A–B). Mutations in key genes such as APC and TP53 displayed distinct patterns between the two risk subgroups, with APC mutations being slightly more frequent in the low-risk group (75% vs. 69%) and TP53 mutations more prevalent in the high-risk group (60% vs. 50%). These genetic alterations contribute to three major oncogenic pathways: WNT–β-catenin signaling, genome integrity maintenance, and the MAPK signaling pathway. Notably, the high-risk subgroup exhibited a higher overall mutation burden within the genome integrity pathway (79% vs. 66%), highlighting pathway-specific vulnerabilities that may underlie differences in tumor behavior and therapeutic response.
Fig. 3
Genomic Alterations Characterizing PRG Subtypes in Colorectal Cancer. A-B Oncoplots highlighting the most frequently mutated oncogenic pathways and their representative genes within each PRG-defined subtype. Panel A: High-risk PRG subtype; Panel B: Low-risk PRG subtype. C-D Copy number alteration frequency plots, displaying the distribution of genomic amplifications (red) and deletions (blue) across PRG subtypes. Panel C: High-risk PRG subtype; Panel D: Low-risk PRG subtype. E-H Results of GISTIC 2.0 analysis showing significant focal copy number alterations within PRG subtypes, with amplified regions marked in red (Panel E: High-risk PRG subtype; Panel F: Low-risk PRG subtype) and deleted regions in blue (Panel G: High-risk PRG subtype; Panel H: Low-risk PRG subtype), along corresponding chromosomal loci
We observed subtle but meaningful differences in the frequency and distribution of copy number alterations (CNAs) between the PRG subtypes (Fig. 3C-D). The high-risk subgroup exhibited a slightly elevated burden of both amplifications and deletions across the genome (Fig. 3C). Specifically, deletions on chromosomes 1 and 4 were more pronounced in the high-risk group, whereas the low-risk subgroup showed increased deletions on chromosomes 5 and 11 (Fig. 3D). Notably, amplification events on chromosome 8 were more prominent in the high-risk subgroup, suggesting potential oncogene-driven genomic instability. Subgroup-specific CNAs revealed distinct genomic signatures (Fig. 3E–H). Amplifications on chromosomes 1, 3, 7, 10, 18, and 21 were uniquely enriched in the high-risk subgroup, while chromosomes 5, 19, and 22 showed amplifications specific to the low-risk subgroup. Similarly, unique deletions were detected on chromosomes 12 and 13 in the high-risk group, and on chromosomes 9 and 17 in the low-risk group. These alterations may reflect differential genomic evolution and contribute to the distinct biological behaviors of each subtype.
Transcriptomic Differences between PRG SubtypesThe identified PRG subtypes exhibited distinct clinical, molecular, and mutational characteristics. To further explore the underlying biological mechanisms differentiating these subtypes, we conducted a transcriptomic analysis to uncover key functional pathways. The high-risk subgroup shows enhanced activation of pathways involved in epithelial to mesenchymal transition (EMT), angiogenesis, and signaling pathways such as tumor growth factor beta (TGF-β), Notch, and Hedgehog, indicating a more invasive, stem-like, and immune-evasive phenotype (Fig. 4A). In contrast, the low-risk subgroup is marked by upregulation of metabolic (oxidative phosphorylation, lipid metabolism, peroxisome) and proliferative (G2M checkpoint, E2F targets) pathways, reflecting a more metabolically active but less aggressive tumor behavior. In the PRG subtypes, distinct metabolic profiles were observed (Fig. 4B). The low-risk subgroup displayed broad enrichment in diverse metabolic pathways, including nucleotide metabolism, amino acid degradation, lipid metabolism, vitamin and drug metabolism, and glycan biosynthesis, reflecting a more active but balanced metabolic state. In contrast, the high-risk subgroup showed selective upregulation of glycosaminoglycan biosynthesis (chondroitin sulfate), glycosphingolipid biosynthesis (ganglio series), and phenylalanine metabolism, pointing to enhanced glycan remodeling and amino acid metabolism associated with tumor aggressiveness.
Fig. 4
Transcriptomic alterations between PRG subtypes.A Heatmap illustrating the differential activity of Cancer Hallmark pathways across PRG subtypes, as determined by single-sample gene set enrichment analysis (ssGSEA). (Wilcoxon rank-sum test; *P < 0.05; **P < 0.01; ***P < 0.001). B Heatmap showing correlations between KEGG metabolic pathways and Cancer Hallmark pathways among PRG subtypes. (Wilcoxon rank-sum test; *P < 0.05; **P < 0.01; ***P < 0.001). C Bubble plot representing Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) between PRG subtypes. D Bubble plot showing KEGG pathway enrichment analysis based on DEGs between PRG subtypes
Complementary Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses based on differentially expressed genes (DEGs = 27; logFC ≥ 0.5 and an adjusted p-value < 0.05) further supported the aforementioned biological implications (Fig. 4C-D). GO terms indicated enhanced regulation of hormone metabolism, oxidative stress response, and ion transport, along with modulation of non-canonical Wnt signaling (Fig. 4C). KEGG pathways highlighted enriched immune-related signaling, hormone secretion, and metabolic processing, suggesting that the transcriptional differences between subgroups are linked to variations in immune activity, metabolic regulation, and hormonal control (Fig. 4D).
The PRG Subtypes are Primarily Distinguished by Differences in Stromal FeaturesNext, we investigated the differences in tumor microenvironment (TME) modulation between the two PRG subtypes. To explore this, we first assessed the immune and stromal components using the ESTIMATE (Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data) algorithm, a computational method used to infer the presence of stromal and immune cells in tumor samples using gene expression data. This analysis revealed that the most notable distinction between the subgroups lay in the stromal features, with significant enrichment observed in the PRG high-risk subgroup (Fig. 5A-C). Further analysis of immune cell composition using the CIBERSORT (Cell-type Identification By Estimating Relative Subsets Of RNA Transcripts) algorithm showed no substantial overall differences in immune infiltration between the subgroups, apart from a few specific cell types, including plasma cells, resting memory CD4 + T cells, undifferentiated macrophages (M0), and activated dendritic cells (Fig. 5D). Notably, M0 macrophages were significantly elevated in the high-risk subgroup. To further dissect stromal differences, we applied the MCP-counter (Microenvironment Cell Populations counter) algorithm, which identified a significant increase in monocytic lineage cells, endothelial cells, and fibroblasts within the PRG high-risk subgroup (Fig. 5E). These cell types not only showed greater abundance but also positively correlated with the PRG risk score (Fig. 5F-K). Importantly, fibroblast enrichment was particularly reflective of PRG risk stratification. Among the PRG risk genes, those upregulated in the high-risk subgroup (e.g., ABCD1, FNDC5, HAO2, PEX5L) showed strong positive correlations with fibroblast presence, whereas downregulated genes (ABCD3, ACOX1, NOS2) were negatively correlated, underscoring the stromal-driven heterogeneity between the PRG subtypes (Fig. 5L).
Fig. 5
Immune and Stromal Landscape of PRG Subtypes. A-C Bar plots comparing ESTIMATE score, immune score, and stromal score across PRG subtypes, reflecting variations in the tumor microenvironment (TME). (Wilcoxon rank-sum test; *P < 0.05; **P < 0.01; ***P < 0.001). D Bar plot of immune cell composition (22 subtypes) across PRG subtypes as assessed by the CIBERSORT algorithm (Wilcoxon rank-sum test; *P < 0.05; **P < 0.01; ***P < 0.001). E Heatmap displaying immune and stromal cell infiltration profiles across TMET subtypes based on MCP-counter analysis. F-H Violin plots illustrating differences in infiltration of monocytic lineage cells, endothelial cells, and fibroblasts between PRG subtypes. (Wilcoxon rank-sum test; *P < 0.05; **P < 0.01; ***P < 0.001). I-K Scatter plots showing correlations between riskScore and infiltration levels of monocytic lineage cells, endothelial cells, and fibroblasts. (Spearman’s correlation test; *P < 0.05; **P < 0.01; ***P < 0.001). L Heatmap presenting the correlation between individual PRG risk genes and infiltration of monocytic lineage cells, endothelial cells, and fibroblasts (Spearman’s correlation test; *P < 0.05; **P < 0.01; ***P < 0.001)
Single-Cell Validation of PRG Risk GenesTo validate the cellular expression patterns of PRG risk genes, we performed single-cell transcriptomic analysis using the colorectal cancer dataset EMTAB8107 (7 patients, 23,176 cells) from the TISCH (Tumor Immune Single-cell Hub) database. t-SNE analysis identified 20 clusters representing 12 distinct cell types, which were categorized into tumor, immune, and stromal compartments of the TME (Fig. 6A–C). Visualization of the PRG risk score via t-SNE and violin plots demonstrated higher expression in malignant (malignant and epithelial) and stromal (endothelial and fibroblast) cells compared to immune cells (Fig. 6D–E). At the gene level, ABCD3 and ACOX1 showed predominant expression in malignant and epithelial cells, while ABCD1 and FNDC5 were also enriched in stromal cells, including fibroblasts, endothelial cells, and monocytes/macrophages (Fig. 6F). Notably, the expression levels of ABCD3 and ACOX1 were lower in malignant cells compared to epithelial cells, which may suggest a progressive downregulation during malignant transformation. The predominant expression of ABCD3 and ACOX1 in this dataset suggests that the patients included may correspond to the low-risk PRG subtype.
Fig. 6
Single-Cell Landscape of PRG Risk Gene Expression in Colorectal Cancer. A-B t-SNE plots showing cellular clustering and annotation of major cell types in the CRC single-cell dataset (EMTAB8107). C t-SNE plot highlighting the distribution of key cellular lineages. D-E Visualization of PRG score expression across cell types using t-SNE and violin plots. (M-N) Bubble plots illustrating the expression patterns of individual PRG risk genes across various cell types in EMTAB8107
FNDC5 and ACOX1 as Representative Biomarkers of PRG-Based SubtypesTo differentiate the PRG-based subtypes, we next aimed to identify robust representative biomarkers using a random forest classifier. Initially, diagnostic feature genes were selected based on classification importance. The MeanDecreaseGini metric from the diagnostic random forest analysis revealed FNDC5 and ACOX1 as the top two features, indicating their highest contribution to sample classification (Fig. 7A-B). Subsequently, univariate Cox regression and random survival forest analysis were performed to identify prognostic biomarkers associated with overall survival (Fig. 7C-E). Both FNDC5 and ACOX1 again demonstrated the highest variable importance scores in the random survival forest analysis, highlighting their critical role in prognostic predictions for the PRG-based risk subgroups.
Fig. 7
FNDC5 and ACOX1 Identified as Key Biomarkers for PRG-Based Subtypes.A Classification error rate plot from the diagnostic random forest model, showing model performance with increasing number of trees. B Feature importance of genes ranked by MeanDecreaseGini from the diagnostic random forest, identifying FNDC5 and ACOX1 as top contributors. C Error rate plot from the random survival forest analysis used to assess prognostic relevance. D Out-of-bag variable importance scores from the survival forest model, highlighting FNDC5 and ACOX1 as leading prognostic markers. E Bar plot showing relative importance of prognostic variables, filtered to those with relative importance > 0.5. F Bar plot illustrating the distribution of COAD patients and their clinical features across four subgroups based on median expression of FNDC5 and ACOX1: FNDC5-high, both-high, ACOX1-high, and both-low. G Principal component analysis (PCA) plot of all four expression-based subgroups, with PC1 explaining 54.5% of the variance. H PCA of FNDC5-high and ACOX1-high subgroups only, with PC1 accounting for 76% of the variance. I ROC curves evaluating the discriminatory power of FNDC5 and ACOX1 between PRG high- and low-risk subgroups
To further assess their relevance, we categorized COAD patients based on the median expression levels of these two genes, leading to four subgroups: FNDC5-high, both-high, ACOX1-high, and both-low (Fig. 7F). The PRG low-risk subgroup primarily consisted of ACOX1-high patients, while the PRG high-risk subgroup was more heterogeneous, comprising the remaining three expression combinations. Principal component analysis (PCA) indicated that PC1 explained 54.5% of the variance across all four subgroups, and 76% when only the FNDC5-high and ACOX1-high subgroups were considered (Fig. 7G-H). Additionally, ROC analysis demonstrated that ACOX1 achieved an AUC of 0.722 and FNDC5 achieved an AUC of 0.807 in distinguishing between the PRG risk subgroups (Fig. 7I). These findings suggest that FNDC5 and ACOX1 serve as promising biomarkers for defining PRG-based risk subgroups, offering potential for improving prognostic stratification.
Experimental Validation of FNDC5 in the Proliferation of colon Cancer CellsTo experimentally validate candidate biomarkers, we selected FNDC5, identified as a marker of the PRG high-risk subgroup, given its previously unexplored role in colon cancer compared to ACOX1, which has been reported to exert a protective effect [28]. Analysis of mRNA expression levels revealed that all PRG risk genes including FNDC5 were upregulated in colon cancer cell lines (SW480 and HCT116) compared to normal colon cells (FHC), with exception of HAO2 (Fig. 8A). NOS2 was excluded from qPCR analysis because its expression requires induction by TNF-α treatment in cancer cells [29]. FNDC5 was prioritized for further investigation due to its strong specificity for PRG subgroup differentiation and prognostic relevance in colon cancer. Western blot analysis confirmed a significant increase in FNDC5 protein expression in cancer cells (p < 0.001) (Fig. 8B-C). To explore its functional role, FNDC5 expression was silenced in SW480 and HCT116 cells using siRNA transfection. Both qPCR and western blot assays validated the efficient knockdown of FNDC5 expression (Fig. 8D-G). Functional assays demonstrated that FNDC5 silencing markedly reduced COAD cell viability in both cell lines, as shown by CCK-8 assays (Fig. 8H-I), and significantly impaired cell proliferation, as evidenced by colony formation assays (Fig. 8J-K).
Fig. 8
Experimental validation of FNDC5 in colon cancer cell proliferation. A Relative mRNA expression levels of PRG risk genes in normal colon cells (FHC) and colon cancer cells (SW480 and HCT116) as assessed by qualitative PCR. The data represent the mean ± SEM (standard error of mean) of n = 3 independent experiments (independent biological replicas) for each condition. Two-tailed unpaired T-test; *P < 0.05; ****P < 0.0001; ns, not significant. B Western blot analysis showing FNDC5 protein expression in FHC, SW480, and HCT116 cells. C Bar plot comparing FNDC5 protein levels (from western blot results) between FHC, SW480, and HCT116 cells. Data are mean ± SEM from n = 3 independent experiments. Two-tailed unpaired t-test: *P < 0.05, **P < 0.0001, ns = not significant. D-G qPCR and western blot analysis of FNDC5 expression in SW480 and HCT116 cells transfected with two different FNDC5 siRNAs. Data are mean ± SEM from n = 3 independent experiments. Two-tailed unpaired t-test: *P < 0.05, **P < 0.0001, ns = not significant. H-I CCK-8 assay showing the effects of FNDC5 silencing (siRNA2) on cell viability in SW480 and HCT116 cells after 48 h of transfection. (One-way ANOVA followed by Dunnett’s post‐test; *P < 0.05, **P < 0.01; ***P < 0.001). J-K Colony formation assay showing the effects of FNDC5 silencing (siRNA2) on colony-forming ability of SW480 and HCT116 cells after 48 h of transfection. Data are presented as means ± standard deviation (SD), n = 3; *P < 0.05, **P < 0.01
Validation of FNDC5 Expression in Clinical Colon Cancer SpecimensWe further validated FNDC5 expression in clinical specimens from colon cancer patients (n = 15) using immunohistochemistry (IHC). These samples were obtained from the Department of Radiation Oncology, Guangzhou Institute of Cancer Research, Affiliated Cancer Hospital of Guangzhou Medical University, Guangzhou, China. Representative IHC images illustrate the distinct expression patterns of FNDC5, as shown in Fig. 9A. FNDC5 was significantly upregulated in CRC tissues compared to adjacent normal tissues (p < 0.001) (Fig. 9B).
Fig. 9
Validation of FNDC5 expression in colorectal cancer. A-B Representative immunohistochemistry (IHC) images and corresponding bar plot illustrating the differential protein expression of FNDC5 in normal versus colorectal cancer tissues (n = 15). Magnification: left panel, 100 μm; right panel, 40 μm. Statistical significance is denoted as *P < 0.05, **P < 0.01, ***P < 0.001
Independent Validation of Prognostic PRG Signatures in Colorectal CancerTo externally validate the prognostic relevance of the identified peroxisome-related risk genes (ABCD1, ABCD3, ACOX1, FNDC5, HAO2, NOS2, and PEX5L), we utilized the Kaplan-Meier Plotter platform for colorectal cancer (https://kmplot.com/), which integrates expression and survival data from 15 independent GEO datasets. Kaplan-Meier survival analyses were conducted for both recurrence-free survival (RFS; n = 1,336) and overall survival (OS; n = 1,061). The results consistently supported our findings: high expression of risk-associated genes (FNDC5, ABCD1, HAO2, and PEX5L; Fig. 10A-H) correlated with significantly poorer RFS and OS, while elevated levels of protective genes (ACOX1, ABCD3, and NOS2) were indicative of favorable outcomes (Fig. 10I-N). These validations reinforce the prognostic utility of the PRG signature across diverse patient populations.
Fig. 10
External Validation of the Prognostic Impact of PRGs. Kaplan-Meier survival curves illustrating differences in recurrence-free survival (RFS, n = 1,336) and overall survival (OS, n = 1,061) between CRC patients with high and low expression of FNDC5 (A-B), ABCD1 (C-D), HAO2 (E-F), PEX5L (G-H), ACOX1 (I-J), ABCD3 (K-L), and NOS2 (M-N)
Comments (0)