
Remember me



In this study, whole-genome sequencing data from lung tissues of seven healthy individuals and ten patients with silicosis (HRA000560)24 were obtained from the Genome Sequence Archive (GSA) database (https://ngdc.cncb.ac.cn/gsa-human/). The sequencing data sources for various stages of silicosis in mouse lung tissues have been previously reported.57 A search of the Gene Set Enrichment Analysis (GSEA) database (http://www.gsea-msigdb.org/) using the keywords “silicosis-associated inflammation and fibrosis genes” identified 201 inflammation-related and 307 fibrosis-related genes. The sequencing data for silicosis patients and mouse lung tissues, along with the key gene lists, are detailed in Supplementary Table S1. In addition, RNA-sequencing data of BALF samples from 176 patients with IPF,58 along with their corresponding prognostic information, were downloaded from the GEO database (http://www.gsea-msigdb.org/) (Supplementary Table S6).
Acquisition of human lung tissue and BALF samplesLung tissue samples from silicosis patients and controls were collected between January 2016 and April 2023 at the Thoracic Surgery Department of the Affiliated Tumor Hospital of Anhui University of Science and Technology (Huainan Oriental Cancer Hospital). The silicosis group comprised three males, while the chronic obstructive pulmonary disease (COPD) group included one female and two males. Bronchoalveolar lavage fluid (BALF) samples were obtained from six male normal miners and ten male silicosis patients between January 2019 and June 2023 at the Huainan Huaihe Energy Group Occupational Disease Prevention and Control Hospital. At the same time, lung function data were collected from 10 male patients with silicosis. During sample collection, each collection provided 100–200 ml of fluid. Lung tissues were rapidly frozen in liquid nitrogen and stored at −80 °C. BALF samples were filtered through sterile gauze, centrifuged, and stored at −80 °C. The use of patient samples in this study was approved by the Ethics Review Committee of Anhui University of Science and Technology (Approval No. HX-001). All participants provided written informed consent, and the study adhered to the principles of the Helsinki Declaration.
Immunofluorescence staining of tissues and cellsImmunofluorescence staining was conducted as previously described.59 In brief, human and murine lung tissues were fixed with 10% formaldehyde, embedded in paraffin, and sectioned into 5 µm slices. After dehydration, antigen retrieval was performed by boiling the sections in EDTA antigen retrieval solution (Servicebio, Cat. No. G1207) at pH 8.0 for 8 min. Sections were outlined with immunohistochemistry pens (Servicebio, Cat. No. G6100) to prevent antibody loss. Following a 30-min block with BSA (Servicebio, Cat. No. GC305010), primary antibodies were applied and incubated overnight at 4 °C, followed by a 1-h incubation with corresponding secondary antibodies at room temperature. Cell nuclei were counterstained with DAPI (Servicebio, Cat. No. G1012) for 10 min in the dark at room temperature. Anti-fluorescence quenching reagent (Servicebio, Cat. No. G1221) was applied for 5 min at room temperature before observation and imaging with a fluorescence microscope (Leica, DMI3000B). Image analysis was performed using ImageJ software (V1.8.0). For cellular immunofluorescence, cells were fixed with 4% formaldehyde for 30 min at room temperature, permeabilized with 0.5% Triton X-100 (Beyotime, Cat. No. P0096), and then incubated with primary and secondary antibodies. The following antibodies were used: PRDX4 (1:200, ab59542, Abcam), PTEN (1:200, A11193, Abclonal), KDEI (1:200, ab176333, Abcam), CD68 (1:500, GB14043, Servicebio), CD80 (1:200, A16039, ABclonal), CD206 (1:500, GB113497, Servicebio), F4/80 (1:1000, GB113373, Servicebio), α-SMA (1:200, ab5694, Abcam), FAM13A (1:200, 55401-1-AP, Proteintech), CD3 (1:100, GB13014, Servicebio), CD16 (1:200, A23541, ABclonal), LY6G (1:400, GB11229, Servicebio), Phospho-NF-kB p65 (1:200, AP0123, ABclonal), c-Jun (1:200, GB11515, Servicebio), Phospho-Akt (Thr308) (1:200, 13038T, CST), and secondary antibodies including HRP-conjugated Goat Anti-Rabbit IgG (1:500, GB23303, Servicebio), FITC-labeled Goat Anti-Rabbit IgG (1:500, GB22303, Servicebio), Cy5-conjugated Goat anti-Rabbit IgG (1:500, GB27303, Servicebio), Cy5 Goat anti-Mouse IgG (1:500, GB27301, Servicebio), Cy3-labeled Goat Anti-Rabbit IgG (1:500, GB21303, Servicebio), Alexa Fluor® 488-conjugated Goat Anti-Mouse IgG (H+L) (1:500, GB25301, Servicebio).
Mouse pulmonary function testingMouse pulmonary function was evaluated using a whole-body plethysmography system (Tow-in, WBP-4M). Mice were acclimated to the testing environment for 30 min before the experiments. Once respiration was stabilized, data collection proceeded for 15 min. The parameters measured included inspiratory time (Ti, seconds), expiratory time (Te, seconds), peak inspiratory flow (PIF, ml/s), peak expiratory flow (PEF, mL/s), respiratory frequency (F, breaths/min), tidal volume (TV, mL), minute ventilation (MV, mL), accumulated volume (AV, mL), expiratory flow at 50% of expired volume (EF50, mL/s), end-inspiratory pause (EIP), end-expiratory pause (EEP), relaxation time (TR), and enhanced pause (PENH). In this study, PIF, PEF, TV, MV, EF50, and PENH were selected as the primary indices of pulmonary function.60,61
Flow cytometric sorting of lung tissuesFor the detection and sorting of cells (Mø, T and NEU) in mouse lung tissues, fluorescent dye-coupled monoclonal antibody (Abs) staining was used for cell surface labeling, including Apc-Cy7 CD45 (BD Pharmingen™, Cat.No 557659, 30-F11, 1/200 dilution), PerCP-Cyanine5.5-CD3e (BD Pharmingen™, Cat.No 551163, 145-2C11, 1/200 dilution), PE-F4/80 (BD Pharmingen™, Cat.No 565410, T45-2342, 1/200 dilution), FITC-CD11b (BD Pharmingen™, Cat.No 557396, M1/70, 1/200 dilution), APC-Ly6G (BD Pharmingen™, Cat.No 560599, 1A8, 1/200 dilution). For the isolation of AMs (alveolar macrophages) and IMs (interstitial macrophages) from lung tissue,62 the following antibodies were used: APC-CD64(eBioscience™, Cat.No 17-0641-80, X54-5/7.1, 1/200 dilution), PE-MerTK(eBioscience™, Cat.No 12-5751-80, DS5MMER,1/200 dilution), PerCP-Cyanine5.5-CD11b(eBioscience™, Cat.No 45-0112-80, M1/70, 1/100 dilution), FITC-CD11c(eBioscience™, Cat.No 11-0114-81, N418, 1/100 dilution). All cells were collected and analyzed using BD FACSAria ™ II flow cytometry (BD FACSDiva™ version V8.0) and FlowJo (version V10).
Cell acquisition and culturePeripheral blood mononuclear cell-derived macrophages (PBMC-m) utilized in this study were differentiated from peripheral blood mononuclear cells (PBMCs) obtained from healthy volunteers. Approval from the Ethics Committee of Anhui University of Science and Technology (Approval No. HX-001) was secured before collecting 10 ml of venous blood from each participant, which was then diluted in PBS solution at a 1:1 ratio. The diluted blood was carefully layered onto 5 ml of Ficoll-Paque PLUS solution (Cytiva, Cat. No. 17144002) for mononuclear cell separation. After centrifugation at 4 °C (300 × g) for 20 min, the mononuclear cell layer was carefully aspirated. The cells were then washed and resuspended in complete RPMI 1640 medium (Thermo Fisher Scientific, Cat. No. 12633012), supplemented with 100 ng/ml macrophage colony-stimulating factor (M-CSF) (MCE, Cat. No. P7050A), and cultured for 14 days to facilitate PBMC differentiation into PBMC-m.63 Giemsa staining (Beyotime, Cat. No. P7050A) and CD68 immunofluorescence were employed to evaluate the purity of the PBMC-m cells.
Alveolar macrophages (AMs) were isolated from approximately 200 mL of fresh bronchoalveolar lavage fluid (BALF) derived from healthy miners. The BALF was first filtered through sterile gauze to eliminate large mucus clots, followed by centrifugation at 4 °C at 300 × g for 10 min. The cells were then passed through a sterile 70 µm cell strainer (BD Falcon, Cat. No. 352350), washed, and resuspended in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Cat. No. C11995500BT), supplemented with 100 ng/mL macrophage colony-stimulating factor (M-CSF) to support AMs growth and maintenance. After 7 days, CD68 immunofluorescence was conducted to evaluate the purity of the AMs.
Other cells including mouse alveolar macrophages MH-S (ATCC, CRL-2019), mouse monocyte/macrophage leukemia cell RAW264.7(Pricella, Cat. No. CL-0190), mouse lung epithelial cells MLE12 (ATCC, Cat. No. CRL-2110), human embryonic lung fibroblasts WI-38 (Pricella, Cat. No. CL-0243), and AAV Pro-293T cells (Clontech, Cat. No. 632273) were cultured in high-glucose DMEM medium containing 10% fetal bovine serum (FBS) (Gibco, Cat. No. C0235) and 1% penicillin/streptomycin (P/S) (Beyotime, Cat. No. C0222). Human non-small cell lung cancer cells A549 (Pricella, Cat. No. CL-0016) were cultured in RPMI 1640 complete medium under the same conditions, while human embryonic kidney cells HEK293T (Pricella, Cat. No. CL-0005) were cultured in MEM (Pricella, Cat. No. CM-0001) complete medium. All cells were cultured in a constant temperature incubator (Panasonic) at 37 °C with 5% CO2. Cell lines used in this study (except for AMs and PBMC-m cells) were tested for mycoplasma contamination and authenticated by STR profiling.
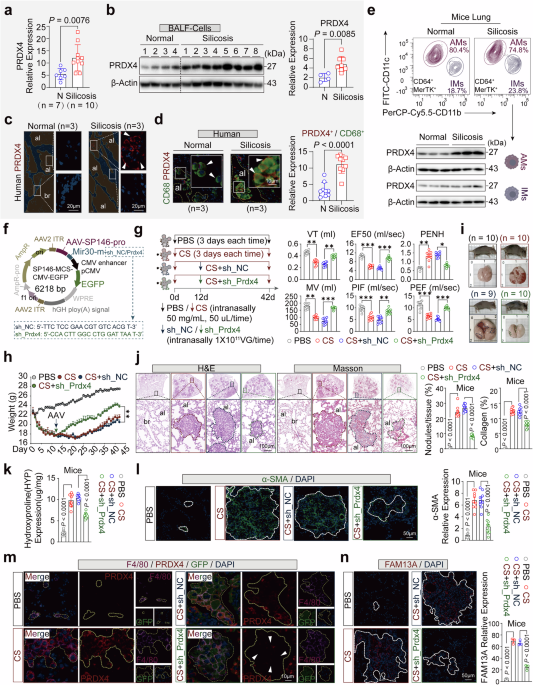
Construction and packaging of the AAV vector for gene interferenceThe interference vector for gene expression, GPAAV-HU6-CMV-eGFP-WPRE, was used as the target gene expression vector, while PHBAAV-RC6 and pHelper served as the packaging plasmids for the AAV9 system. The vector was constructed with a macrophage-specific promoter, SP146 (5-CTAGCGAGGGCGGACCAGAAAAGGAGAAGTAGGAGCCAAGATTTCCAAACTCTGTGGTTGCCTTGCCAAGATTTCCAAACTCTGTGGTTGCCTTGCAGAAAAGGAGAAGTAGGAGAAGCGACTTCCTCTTTCCAGAAGCGACTTCCTCTTTCCAGAGGAAGAGGGCGGAGGCTCACAAGGCAACCACAGAGTTTGGAAATCTTGGAAGCGACTTCCTCTTTCCAGCAGAAAAGGAGAAGTAGGAGAAGCGACTTCCTCTTTCCAGGTCCGCCCTCG-3), and gene sequences targeting PRDX4 based on miR30,64 including sh_NC (5-TTCTCCGAACGTGTCACGT-3) and sh_PRDX4 (5- CCACTTGGCC-TGGATTAAT-3). After digestion with Bsu15I (Thermo Fisher Scientific, Cat. No. ER0141) and EcoRI (Thermo Fisher Scientific, Cat. No. ER0271) enzymes, the target gene fragments were inserted into the interference vector using the Hieff CloneTM recombination reaction system. The ligation products were transformed into DH5α (Sangon, Cat. No. B528413) competent cells, and clones were selected for sequencing at a commercial sequencing company (Sangon) to confirm the sequence identity of the inserted fragments with the designed oligo sequences. The resulting clones were confirmed to be the GPAAV-SP146-sh_NC-CMV-eGFP-WPR control and GPAAV-SP146-sh_Prdx4-CMV-eGFP-WPRE interference vectors. AAV Pro-293T cells were employed as the packaging cells for adeno-associated viruses, and the E. coli strain stbl3 was used for amplifying the viral vector and helper packaging plasmids (PHBAAV-RC6 and pHelper). Plasmid extraction kits (Qiagen, Cat. No. 12143) were used to extract the successfully constructed adeno-associated virus recombinant plasmids and packaging plasmids, and their concentration and purity were determined by the UV absorption method to ensure that the A260/A280 ratio of the purified plasmid DNA was between 1.8 and 2.0. The day before transfection, when the confluence of the cells in a 10 cm culture dish reached 60% to 80%, the transfection preparation was initiated. One to two hours before transfection, the cells were switched to fresh serum-free DMEM. The HG transgene reagent (Genomeditech, Cat. No. TG-10012) served as the transfection reagent, and the transfection system consisted of DMEM (850 μL) + interference plasmid (10 μg) + PHBAAV-RC6 (10 μg) + pHelper (10 μg) + HG transgene reagent (120 μg). After transfection for 6 to 8 h, the cells were refed with complete DMEM medium, and the Enhancing buffer (100×, 100 μL) was added to facilitate transfection. Cells and cell supernatants were harvested 72 h post-transfection, subjected to three freeze-thaw cycles, and centrifuged to collect the viral supernatant. The viral supernatant was concentrated by adding Benzonase (50 U/mL) and MgCl2 (0.002 M) and incubating at room temperature for 30 min, followed by filtration through a 0.22 μm filter. The virus was purified using iodixanol (Millipore, Cat. No. 1343517) gradients (60%, 40%, 25%, 15%), and collected using Amicon® Ultra 50KD (Millipore, Cat. No. UFC5050) ultrafiltration tubes. The virus titer was determined, and the concentration was above 1.16e + 12 VG/mL. The prepared virus was stored at −80 °C for future use.
Macrophage-stimulated epithelial-fibroblast differentiation modelThe model was divided into two types: macrophage/epithelial (MH-S/MLE12, AMs/A549, and PBMC-m/A549) and macrophage/fibroblast (AMs/WI-38, PBMC-m/WI-38) conversion models. Supernatants from macrophages stimulated with CS were collected by centrifugation at 4 °C, followed by filtration through a 0.45 μm filter. The supernatant was mixed with the culture medium at a 1:1 ratio to culture the epithelial/fibroblast cells. CS (Sigma, No. 637238) was a non-toxic, odorless, white powder of 10–20 nm size, which appeared as a white suspension when dissolved in PBS. It needed to be thoroughly mixed before use and could be directly prepared in the culture medium.
Animal model constructionOur study exclusively examined male mice. It is unknown whether the findings are relevant for female mice. A total of 130 SPF-grade C57BL/6 male mice aged 8–10 weeks old (22–24 g) were purchased from Henan Scrofula Biotechnology Co., Ltd. All experimental procedures were approved by the Ethics Committee of Anhui University of Science and Technology (University Lun No. GZ-2023-041). The mice were housed in the Experimental Animal Center of the School of Medicine, Anhui University of Science and Technology, under the following conditions: temperature of 20 ± 2 °C, humidity of 55 ± 5%, and a 12-h light-dark cycle. They had ad libitum access to drinking water and were fed three times a week with regular food. Daily weight measurements were recorded. After a 3-day adaptation period, the mice were randomly divided into different groups to construct the animal models.
In the silica-induced lung fibrosis mouse model with PRDX4 knockdown in lung AMs, four experimental groups were established: PBS (n = 10), CS (n = 10), CS+sh_NC (n = 9), and CS+sh_Prdx4 (n = 10). Mice in the PBS and CS groups were administered intranasal PBS (50 μL) or 50 μL CS (50 mg/ml) every three days for 42 days. The CS+sh_NC and CS+sh_Prdx4 groups received a single intratracheal instillation of 50 μl sh_NC/Prdx4 viral suspension (1 × 1011 VG) on day 12. CS (Sigma, Cat. No. S5631), a non-toxic and odorless white powder with particle sizes between 0.5 and 10 μm, formed a milky white suspension when mixed with PBS. Daily monitoring of mouse body weight and survival was conducted, with the exception of one mouse in the CS+sh_NC group that died accidentally during anesthesia. At the conclusion of the 42-day study period, the mice were euthanized, and lung function was evaluated at both the start and finish.
In the Conoidin A (Con A) treatment study of CS-induced silicosis mouse models at the inflammatory phase (day 21), transitional phase (day 42), and fibrotic phase (day 63), the animals were divided into seven groups: PBS group (n = 10), CS-21 group (n = 10), CS-21 + Con A group (n = 10), CS-42 group (n = 10), CS-42 + Con A group (n = 10), CS-63 group (n = 10), and CS-63 + Con A group (n = 10). Mice in the PBS and CS groups received intranasal instillation of 50 μL PBS or 50 μL CS suspension (50 mg/ml) every three days, for a total duration of 21, 42, or 63 days, respectively. Concurrently, Con A (5 mg/kg) was administered via intraperitoneal injection at a volume of 50 μL every three days. Con A was prepared as a clear solution with 10% DMSO, 40% PEG300, 5% Tween-80, and 45% Saline.
In the CS-induced silicosis model in which Con A treatment was initiated after fibrosis formation (day 42), mice were divided into two groups: CS-42 + DMSO-21 group (n = 10) and CS-42 + Con A-21 group (n = 10). All mice received intranasal instillation of 50 μL CS suspension (50 mg/ml) every three days for 42 days, after which CS administration was discontinued. Subsequently, mice in the CS-42 + Con A-21 group were administered Con A (50 μL, 5 mg/kg) via intraperitoneal injection every three days, while the CS-42 + DMSO-21 group received DMSO as a control. Con A administration was stopped after 21 days of treatment (day 63). Daily monitoring of mouse body weight and survival was conducted, and lung function was assessed at the study’s commencement and conclusion. The study adhered to stringent animal welfare principles, including twice-daily evaluations of mouse appetite, activity, behavior, body temperature, and body weight changes. Euthanasia was performed at the study’s end using 1% isoflurane (RWD, Cat. No. S5631) followed by cervical dislocation.
H&E, Masson staining, Sirius red stain, and HYP testingAs described previously,59 tissues were fixed with formalin, dehydrated, and embedded in paraffin for subsequent staining with hematoxylin and eosin (H&E), Masson’s trichrome, and Sirius red stain. For H&E staining, lung tissue sections were deparaffinized and rehydrated using xylene and a graded series of ethanol (100%, 90%, 80%, and 70%). The sections were stained with hematoxylin solution (Beyotime, Cat. No. C0105S) for 3–5 min at room temperature, followed by rinsing in running water for 5 min to remove excess staining. Then, the sections were stained with eosin staining solution for 1 min, followed by two washes with 70% ethanol. After mounting with neutral gum (Solarbio, Cat. No. G8590), the sections were observed and photographed using an optical microscope (OLYMPUS, BX53 + DP74). Cell nuclei appeared blue, while the cytoplasm appeared pink or red. For Masson’s trichrome staining, lung sections were deparaffinized following the same procedure as described above. The sections were stained with Weigert’s iron hematoxylin solution (Beyotime, Cat. No. C0189S) for 10 min at room temperature, rinsed in running water for 5 min to remove excess staining, stained with Ponceau acid fuchsin staining solution for 10 min at room temperature, rinsed in running water for 10 s, differentiated in phosphomolybdic acid solution for 2 min, and then rinsed. Finally, the sections were stained with aniline blue staining solution for 1 min, rinsed in running water for 10 s, mounted with neutral gum (Solarbio), and observed and photographed using an optical microscope (OLYMPUS). In the trichrome stain, cell nuclei appeared purple, muscle fibers and cytoplasm appeared red, and collagen fibers appeared blue. ImageJ software (V1.8.0) was used to analyze the number of lung lobes and the level of fibrosis. For Sirius Red staining, lung tissue sections were deparaffinized following the standard procedure described above. Sections were then stained with Weigert’s iron hematoxylin solution for 5–10 min, followed by a brief rinse in distilled water for 10–20 s to remove excess stain. Sections were washed under running tap water for 5 min. Subsequently, sections were stained with Sirius Red solution (Servicebio, #G1078) for 15–30 min, then rinsed thoroughly under running water to remove surface dye. Dehydration was performed sequentially in 75% ethanol for 1 min, 95% ethanol for 1 min, and absolute ethanol for 1 min, followed by three changes of xylene, each for 1–2 min. Finally, sections were mounted with neutral resin (Solarbio) and examined under an optical microscope (OLYMPUS) for imaging. Collagen fibers appeared red, nuclei ranged from brownish to black, and muscle fibers stained yellow. ImageJ software (V1.8.0) was used to analyze the level of fibrosis. For measurement of the hydroxyproline (HYP) content in mouse lung tissues using the assay kit (BC0250, Solarbio), approximately 0.2 g of lung tissue was homogenized with 2 mL extraction solution. The mixture was boiled and digested at 16,000 rpm for 2–4 h until no obvious clumps were present. After cooling, the pH was adjusted to 6–8 with 10 mol/L NaOH. The volume was brought to 4 mL with distilled water, and the supernatant was collected after centrifugation at 16,000 rpm for 20 min. A standard solution with a concentration of 0.5 mg/mL was prepared by diluting a series of concentrations. Blank, sample, and standard tubes were prepared. After heating in a water bath at 60 °C for 20 min and standing at room temperature for 15 min, the optical density (OD) was measured at 560 nm. The ∆A (A standard tube - A blank tube) was calculated, and then a standard curve was plotted using the equation y = kX + b. The ∆A value for the test tube was substituted into the equation to calculate X (µg/mL). Finally, the tissue hydroxyproline content (µg/g) was calculated as X × sample volume (V1) / (sample mass (W) × added sample volume (V1) / volume of the tissue extract (V2)).
Real-time quantitative polymerase chain reaction (RT-qPCR)Total RNA was extracted from cells and tissues using Trizol reagent (Thermo Fisher Scientific, Cat. No. 15596026) according to the manufacturer’s instructions. cDNA was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Cat. No. K1622). The resulting cDNA was used for real-time quantitative PCR (RT-qPCR) with the Genious 2× SYBR Green Fast qPCR Mix (ABclonal, Cat. No. RK21204) system for gene amplification. Quantitative analysis was performed using the 2−∆∆Ct method, with GAPDH used as an endogenous reference. Primer design and synthesis were provided by Sangon Biotech.
m_Prdx4: F: TCCTGTTGCGGACCGAATC, R: CCACCAGCGTAGAAGTGGC;
m_Il-1α: F: CGAAGACTACAGTTCTGCCATT, R: GACGTTTCAGAGGTTCTCAGAG;
m_Il-1β: F: GCAACTGTTCCTGAACTCAACT, R: ATCTTTTGGGGTCCGTCAACT;
m_Il-6: F: TAGTCCTTCCTACCCCAATTTCC, R: TTGGTCCTTAGCCACTCCTTC;
m_Tnf-α: F: CCCTCACACTCAGATCATCTTCT, R: GCTACGACGTGGGCTACAG;
m_Tgf-β: F: CTCCCGTGGCTTCTAGTGC, R: GCCTTAGTTTGGACAGGATCTG;
m_α-sma: F: GTCCCAGACATCAGGGAGTAA, R: TCGGATACTTCAGCGTCAGGA;
m_Col1a1: F: GCTCCTCTTAGGGGCCACT, R: CCACGTCTCACCATTGGGG;
m_Col3a1: F: CTGTAACATGGAAACTGGGGAAA, R: CCATAGCTGAACTGAAAACCACC;
m_Gapdh: F: AGGTCGGTGTGAACGGATTTG, R: TGTAGACCATGTAGTTGAGGTCA;
h_PRDX4: F: AGAGGAGTGCCACTTCTACG, R: GGAAATCTTCGCTTTGCTTAGGT;
h_IL-1α: F: AGATGCCTGAGATACCCAAAACC, R: CCAAGCACACCCAGTAGTCT;
h_IL-1β: F: ATGATGGCTTATTACAGTGGCAA, R: GTCGGAGATTCGTAGCTGGA;
h_IL-6: F: ACTCACCTCTTCAGAACGAATTG, R: CCATCTTTGGAAGGTTCAGGTTG;
h_TNF-α: F: GAGGCCAAGCCCTGGTATG, R: CGGGCCGATTGATCTCAGC;
h_TGF-β: F: CAATTCCTGGCGATACCTCAG, R: GCACAACTCCGGTGACATCAA;
h_a-SMA: F: CTATGAGGGCTATGCCTTGCC, R: GCTCAGCAGTAGTAACGAAGGA;
h_COL1A1: F: GAGGGCCAAGACGAAGACATC, R: CAGATCACGTCATCGCACAAC;
h_COL3A1: F: TTGAAGGAGGATGTTCCCATCT, R: ACAGACACATATTTGGCATGGTT;
h_GAPDH: F: ACAACTTTGGTATCGTGGAAGG, R: GCCATCACGCCACAGTTTC.
Design of mutant site and construction of overexpressed plasmidProtein site-directed mutagenesis and plasmid construction were performed as described previously.59 In summary, based on the principles of amino acid site mutation, the mutated amino acids needed to best preserve the structure and function of the original protein, considering factors such as atomic composition, polarity, charge, and size. Using the disease mutation sites and protein structure visualization tool VarSite (https://www.ebi.ac.uk/thornton-srv/databases/VarSite),41 simulations were performed to mutate four cysteine residues (51, 124, 148, and 245) of the PRDX4 protein (Uniprot ID: Q13162). The impact of the mutated amino acids on the structure and function of the PRDX4 protein was simulated and analyzed, and mutation sites that had minimal impact on the protein structure and function were selected. The results showed that the optimal mutations for Cys (51, 124, and 245) were to serine (S), and the optimal mutation for Cys148 was to aspartic acid (D). Based on these optimal mutation sites, the wild-type plasmid H_PRDX4 WT, mutant plasmids H_PRDX4 (p.C51S) Mut1, H_PRDX4 (p.C124S) Mut2, H_PRDX4 (p.C148D) Mut3, and H_PRDX4 (p.C245S) Mut4 were constructed. The pcDNA3.1-MCS-EF1-ZsGreen plasmid (Genomeditech, Cat. No. GM-8630P1) was used as the destination gene expression vector, and the E. coli strain stbl3 (Thermo Fisher, Cat. No. G1036) was prepared for transformation. PRDX4 primers were designed and synthesized with the homologous recombination sequence added at the 5′ end. The designed primer sequences were sent to a primer synthesis company (Suzhou Jinweizhi Biotechnology Co., Ltd.) for synthesis. After plasmid digestion, Sanger sequencing validation, and extraction (TIANGEN, Cat. No. DP107), the pcDNA3.1-H_PRDX4-3×HA-EF1-ZsGreen1WT, pcDNA3.1-H_PRDX4(p.C51S)-3×HA-EF1-ZsGreen1 Mut1, pcDNA3.1-H_PRDX4(p.C124S)-3×HA-EF1-ZsGreen1 Mut2, pcDNA3.1-H_PRDX4(p.C148D)-3×HA-EF1-ZsGreen1 Mut3, and pcDNA3.1-H_PRDX4(p.C245S)-3×HA-EF1-ZsGreen1 Mut4 overexpression plasmids were obtained. Using pcDNA3.1-MCS-EF1-ZsGreen as the destination gene expression vector plasmid, the steps were the same as mentioned earlier, and the pcDNA3.1-H_PTEN-3×Flag-EF1-ZsGreen1WT wild-type overexpression plasmid was constructed.
Plasmid and siRNA transfectionIn plasmid transfection, HEK293T cells (Pricella) were seeded the day before transfection in a 60 mm dish (Pricella), with the cell density reaching 60% to 80% for transfection. Six hours prior to transfection, the cells were switched to non-complete MEM (Pricella) medium. The plasmid (2 μg) was diluted in 400 μl of Opti-MEM™ I (Thermo Fisher Scientific, Cat. No. 31985070), and Lipofectamine™ 2000 (Thermo Fisher Scientific, Cat. No. 11668500) was diluted in another 40 μl of Opti-MEM™ I (8 μl). After mixing the solutions gently, the Lipofectamine™ 2000 mixture was slowly added to the plasmid mixture, incubated for 10 min at room temperature. The prepared transfection solution was then added to the cell culture dish, gently mixed, and the cells were incubated at 37 °C with 5% CO2 in a culture incubator (Panasonic) for 6 h. The medium was then replaced with complete MEM, and the expression of green fluorescent protein (ZsGreen) in HEK293T cells was observed under a fluorescence microscope (Leica) 24 h post-transfection to assess plasmid transfection efficiency. In siRNA transfection, the cells were seeded in a 35 mm dish the day before transfection, with the cell density reaching 60–80% for transfection. Six hours prior to transfection, the cells were switched to non-complete RPMI 1640/DMEM medium (without FBS and without P/S). The siRNA (20 pmol) was diluted in 200 μl of serum-free medium, and Lipofectamine™ 2000 (4 μl) was diluted in another 200 μl of serum-free medium. The siRNA and Lipofectamine™ 2000 solutions were mixed and added to the cells in the same manner as for plasmid transfection. After incubation for 6 h at 37 °C with 5% CO2 in a culture incubator, the medium was replaced with complete RPMI 1640/DMEM. The expression levels of the target gene and protein were detected by RT-QPCR and WB at 24 and 48 h post-transfection, respectively. The siRNAs and their corresponding negative controls used in this study were purchased from Genepharma (Shanghai, China). The siRNA sequences for mouse cell lines were as follows: si_NC: ACGUGACACGUUCGGAGAATT, si_Prdx4_573: AUUAAUCCAGGCCAAGUGGTT. For human cell lines, the siRNA sequences were: si_NC: UUCUCCGAACGUGUCACGUTT, si_PRDX4_531: CUGGCAGUGACACGAUUAATT.
Reducing and non-reducing SDS-PAGEReducing and Non-Reducing SDS-PAGE were performed as previously described.59,65 Briefly, treated cells were lysed in RIPA buffer containing 1×PMSF (100 mM) (Beyotime, Cat. No. ST506) and 20 mM N-Ethylmaleimide (NEM) (Sigma, Cat. No. E376) on ice for 30 min. After centrifugation at 4 °C for 30 min, the supernatant was collected and the protein concentration was determined using a BCA protein quantification kit (Thermo Scientific Pierce, Cat. No. A55864). For the separation of cytoplasmic and nuclear proteins, the NE-PER™ nuclear and cytoplasmic extraction kit (Thermo Scientific Pierce, Cat. No. 78833) was utilized. In Reducing SDS-PAGE, an equal amount of protein was mixed with 5× SDS-PAGE loading buffer (Beyotime, Cat. No. P0015L) and boiled at 100 °C for 10 min. Thiol-reducing agent TCEP (5 mM) (Beyotime, Cat. No. ST045) was added and incubated for 30 min prior to separation on 10% NuPAGE™ Tris-Glycine gels (constant voltage of 60 V for 45 min for the stacking gel and 110 V for 1 h for the separating gel). In Non-Reducing SDS-PAGE, an equal amount of protein was mixed with 4× NuPAGE™ LDS sample buffer (Thermo Fisher Scientific, Cat. No. NP0007) and heated at 70 °C for 10 min prior to separation on the same gels. Following separation, all proteins were transferred to 0.2 μm PVDF membranes (Immobilon, Cat. No. ISEQ00010) at room temperature. The membranes were blocked with 5% BSA for 1 h at room temperature. After TBST washing, primary antibodies were added and incubated overnight at 4 °C, followed by secondary antibodies at room temperature for 1 h (Millipore, Cat. No. WBKLS0100, incubated for 30 s). Protein bands were visualized using an imaging system (Amersham ImageQuant™ 800, 29399481), and band intensities were analyzed using ImageJ software (V1.8.0). The following antibodies were used: PRDX4 (1:1000, ab59542, Abcam), PTEN (1:1000, 9552, CST), β-Actin (1:10,000, AC026, Abclonal), α-SMA (1:1000, 14968, CST), COL1A1 (1:1000, ab138492, Abcam), COL3A1 (1:1000, 30565, CST), E-Cadherin (1:1000, A20798, Abclonal), N-Cadherin (1:500, A3045, Abclonal), Vimentin (1:500, A2584, Abclonal), Phospho-PI3K p85 (1:1000, 4228T, CST), Phospho-Akt (Thr308) (1:1000, 13038T, CST), Phospho-TAK1(Thr184/187) (1:1000, 4508s, CST), Phospho-NF-kB p65 (1:1000, AP0123, ABclonal), Phospho-c-Jun-S73 (1:500, AP0119, ABclonal), HistoneH3 (1:2000, A2348, Abclonal). The corresponding secondary antibodies were HRP-conjugated Goat anti-Rabbit IgG (1:5000, AS014, Abclonal).
ROS detectionAfter PBMC-m and AMs cells were stimulated with CS (50 μg/cm²) for 48 h, DCFH-DA (S0033S, Beyotime) was diluted 1:1000 in serum-free culture medium to a final concentration of 10 μM. Subsequently, 1 mL of the diluted DCFH-DA was added to the 6-well plates and incubated in a cell culture incubator at 37 °C for 20–30 min. After incubation, the cells were washed three times with serum-free culture medium to thoroughly remove DCFH-DA that had not entered the cells. Finally, the cells were observed and imaged using a fluorescence microscope (Leica, DMI3000B).
Recombinant active protein reaction and detectionTo investigate the effects of H2O2 treatment on protein expression, 1 μg of recombinant active PRDX4 (ab93947, Abcam) and PTEN (ab157087, Abcam) proteins were incubated at 37 °C with 100 μM H2O2 for 30 min. The expression of PRDX4 (monomer, dimer, HWM oligomer) and PTEN (monomer, dimer) was detected under both Reducing and Non-Reducing conditions using Coomassie Blue staining. For the detection of PTEN addition post-H2O2 treatment, 1 μg of recombinant active PRDX4 protein was treated with H2O2 for 30 min, followed by the removal of H2O2 and the addition of 1 μg of recombinant PTEN protein, which was then incubated at 37 °C for 2 h. Non-Reducing SDS-PAGE was used to detect the expression of PRDX4 and PTEN proteins under Non-Reducing conditions. To assess the impact of Con A on PRDX4 oligomer formation, 1 μg of recombinant PRDX4 protein was first treated with 100 μM H2O2 for 30 min, followed by the addition of 20 mM Con A (PRDX4 protease inhibitor) and incubation at 37 °C for 30 min. The expression pattern of the PRDX4 protein was detected using Coomassie Blue staining. To evaluate the effect of the sequence of Con A and H2O2 treatment on PRDX4 expression, 1 or 2 μg of recombinant PRDX4 protein was first incubated with 20 mM Con A at 37 °C for 30 min, followed by the addition of 100 μM H2O2 for 30 min, and PRDX4 protein expression was detected using Coomassie Blue staining.
Co-immunoprecipitation assaysImmunoprecipitation experiments were conducted as previously described.23 In brief, 1 × 106 cells were collected by washing with cold PBS (Contains 10 mM NEM) and lysed on ice for 30 min with 1 ml of lysis buffer (Tris-HCl 50 mM, pH 7.4, NaCl 150 mM, Sodium Deoxycholate 0.25%, NP-40 1%, EDTA 1 mM, PMSF 1 mM, 20 mM NEM, 200 U catalase).The cells were then centrifuged at 4 °C at 12600 rpm for 15 min. The supernatant was collected and incubated overnight at 4 °C with the corresponding primary antibody to form the protein-antibody complex. Subsequently, 10 μL of protein A/G agarose beads (Santa Cruz Biotech, Cat. No. sc-2003) were added and incubated for 3 h to deplete endogenous immunoglobulin G and enrich the primary antibody/protein complex. The complex was washed three times with lysis buffer and then incubated with 5× SDS-PAGE sample buffer at 100 °C for 10 min. SDS-PAGE was performed to detect the corresponding protein expression. The protein bands were visualized using an imaging system, and the band intensities were analyzed using ImageJ software (V1.8.0). The following antibodies were used: FLAG (1:100, #2368, CST) and HA (1:1000, #3724, CST).
Endoplasmic reticulum isolationThe endoplasmic reticulum (ER) and cytoplasmic (endoplasmic reticulum-free) fractions of AM cells were isolated using an ER isolation kit (ER0100, Sigma-Aldrich). Briefly, for AMs cells, cells were collected and incubated for 20 min at 4 °C using 1× hypotonic extraction buffer (10 mM HEPES (pH 7.8), 25 mM KCl, and 1 mM EGTA) for 20 min to allow for sufficient expansion and rupture of the cells. centrifugation was performed for 5 min at 600 × g, and then the supernatant was aspirated off, the precipitate was retained, and based on the volume of precipitated PCV cells, 1× isotonic Extraction buffer (10 mM HEPES (pH 7.8), 250 mM sucrose, 25 mM KCl, and 1 mM EGTA) was added, and the cells were broken using a Dounce homogenizer and subjected to differential centrifugation. The homogenate was centrifuged at 1000 × g for 10 min at 4 °C, and the supernatant was collected and the supernatant was centrifuged at 12,000 × g for 15 min at 4 °C, and the supernatant was collected, and this supernatant fraction was the postmitochondrial fraction (PMF). Subsequently, the PMF supernatant was precipitated using CaCl₂ (8 mM), followed by medium-speed centrifugation (8000 × g), and the precipitate was collected to obtain enriched rough endoplasmic reticulum (RER) microsomes.
CIBERSORTx and TRRUST database analysisRNA-seq data from silicosis patients and mouse lung tissues were imported into CIBERSORTx (https://cibersortx.stanford.edu). Subsequently, the “LM22” signature matrix was selected from the “Signature Matrix” dropdown menu to analyze the expression levels of the 22 human immune cell types identified (P < 0.05). The transcription factors (TFs) that regulate human and mouse inflammatory factors TNF-α, TGF-β, IL-1 (IL-1α, IL-1β), and IL-6 were searched using the TRRUST database (http://www.grnpedia.org). Detailed information is provided in Supplementary Table S4.
Statistical analysisStatistical analyses were performed using Rv4.0.2 or GraphPad Prism 9 (GraphPad Software Inc., San Diego, USA). All results were expressed as the mean ± standard deviation (SD). Two-group comparisons were performed using an unpaired, two-tailed Mann–Whitney U-test, and multi-group comparisons were performed using one-way ANOVA with Tukey´s or Bonferroni´s multiple comparison test. Differences were considered statistically significant at P < 0.05. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and NS, not significant.
Comments (0)