
Remember me





DMEM, fetal bovine serum (FBS), and Lipofectamine™ 2000 transfection reagents were purchased from Invitrogen. 1,4-dithiothreitol (DTT), dimethyl sulfoxide (DMSO), Triton X-100, 4% paraformaldehyde, bovine serum albumin, ethanol, Hoechst 33,342, RIPA buffer, phosphatase inhibitor cocktail, all-trans retinoic acid, corn oil, thioflavine S, penicillin, and streptomycin were from Sigma. Blue RangeTM prestained protein molecular marker and BCA Protein Assay Kit were from Thermo Fisher Scientific. Recombinant Human Amyloid Precursor Protein alpha was purchased from Antibodies.com (Cambridge, UK). Anti-ADAM10 antibody (Cat. No. ab124695, 1:5000) and anti-CX3CL1 antibody (Cat. No. ab25088, 1:1000) were purchased from Abcam (Cambridge, UK). Anti-APP antibody (Cat.No.2045, 1:2000) and was purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-6E10 antibody (Cat. No. 803014, 1:5000) was purchased from Biolegend (San Diego, CA, USA). Anti-beta Amyloid antibody (MOAB-2, Cat. No. 13075, 1:500) was purchased from Novus Biologicals (Littleton, CO, USA). Anti-Iba1 antibody (Cat. No. 019-19741, 1:1000) was purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Anti-TREM2 antibody (Cat.No.27599-1-AP, 1:1000) was purchased from Proteintech (Chicago, IL). Anti-CX3CR1 antibody (Cat.No.38,481, 1:1000) was purchased from Signalway Antibody (SAB, College Park, Maryland, USA). HRP-conjugated Affinipure Goat Anti-Mouse IgG (H + L) (Cat. No. SA00001-1, 1:3000), HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H + L) (Cat. No. SA00001-2; 1:3000) were purchased from Proteintech (Chicago, IL). Alexa Fluor 647-conjugated Affinipure Goat Anti-Mouse IgG (H + L) (Cat. No. 115-605-003, 1:1000), Cy3-conjugated AffiniPure F(ab’)2 Fragment Goat Anti-Rabbit IgG (H + L) (Cat. No. 111-166-144, 1:1000) was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). MiR-140-3p and miR-122-5p mimics/ scramble RNA were synthesized by GenePharma (Shanghai, China). The miR-140-3p mimics and miR-122-5p mimics were double-stranded (5ʹ-UACCACAGGGUAGAACCACGG-3ʹ and 5ʹ-UGGAGUGUGACAAUGGUGUUUG-3ʹ, respectively). The scramble RNAs was double-stranded RNA (5ʹ- UUCUCCGAACGUGUCACGUTT-3ʹ).
Participants and sample collectionThe participants, aged between 60-90-year-old, were recruited from the Department of Neurology, the Second Affiliated Hospital of Guangzhou Medical University and the study were approved by the ClinicalTrials.gov (ID: NCT03653156). All patients gave written informed consent. All participants underwent the Mini-Mental State Examination (MMSE) and those scored > = 27 was considered as cognitive normal controls. Participants with a MMSE score < = 26 went through additional tests of cerebrospinal fluid Aβ and p-Tau. Diagnosis of AD was made in accordance with the 2018 NIA-AA Pathological Diagnostic Guidelines. Aβ42 < 550 pg/mL or Aβ42/Aβ40 ≤ 0.05 (Amyloid-beta (1–40) High Sensitive ELISA and Amyloid-beta (1–42) High Sensitive ELISA, Cat. No. RE59781, Cat. No. RE59791, IBL International GmbH., Hamburg, Germany), p-Tau > 50 pg/mL (phosphoTAU ELISA, Cat. No. 30,121,609), and t-Tau > 399 pg/mL (hTau total ELISA, Cat. No. RE59631) was considered ATN positive, and these participants were included in the study providing no presence of other brain diseases. The final cohort consisted of 11 AD and 14 cognitive normal subjects. 2 mL peripheral blood from the cubital vein were collected using EDTA anticoagulation tubes (Cat. No.367863, Becton, Dickinson and Company, Franklin Lakes, NJ, USA) in the early morning after refraining from eating and drinking for eight hours and were allowed to sit for 30 min at room temperature. Plasma samples were collected from the supernatant after centrifugation at 3,000 rpm for 10 min and stored in 100 µL aliquots at -80℃ before use.
Animals4-month-old male APP/PS1 mice and control C57BL/6 mice were purchased from The Experimental Animal Center of Guangdong (Guangzhou, China) and bred in the Laboratory Animal Management Center of the Jinan University in a pathogen free facility. All mice were housed at temperature (21–25℃) under 12 h- light/dark cycle with free access to water and food. All animal experiments were approved by the Institutional Animal Care and Use Committee of Jinan University (IACUC-20230625-02). For atRA treatment, atRA was dissolved in DMSO and diluted in corn oil to make the final concentration is 5% DMSO (v/v). atRA (20 mg/kg) was applied by intraperitoneal injection three times per week. The control mice were mock treated with vehicles. Administration of atRA was initiated 24 h after the stereotactic injection and continued for 8 weeks. All animals were sacrificed using CO2 asphyxiation before the tissue samples were collected.
Stereotactic injectionMice were anesthetized with 2% pentobarbital sodium (50 mg/kg) and placed in the stereotactic platform (Harvard Apparatus, Holliston, MA). AAV9-miR140-miR122-eGFP or AAV9-Scr-eGFP (4 × 1010 genome copies × 2) were injected into both sides of the lateral ventricles (posterior: +0.3 mm; mediolateral: ± 1.0 mm, dorsal: -2.0 mm) at a rate of 0.4 µL/min using a micro-drive arm of the micropump (KDS Legato™ 130 micro-pump, KD Scientific Inc., Holliston, MA) attached to a Hamilton 5 µL syringe (87930 Hamilton, Reno, NV). The needle was held in place for 5 min to prevent leakage before retraction after each injection. Mice were recovered in a warming cabinet before returning to their home cage.
Behavioral assessmentMice were brought to the testing room and allowed to acclimate for 1 h before assessment. A Y-maze apparatus composed of three enclosed arms (35 cm long × 15 cm high × 5 cm wide) oriented at 120° angles from each other was used for the Y maze test to assess short-term spatial working memory. Mice were introduced at the end of one of the arms and allowed for 8-min free exploration. A spontaneous alternation was defined as consecutive entries into three arms. Morris water maze (MWM) test was performed using a circle pool with a diameter of 120 cm diameter and a depth of 60 cm filled with water at room temperature (24℃). The pool was virtually divided into four quadrants and the escape platform (10 cm diameter) was placed in one of the quadrants submerged 1 cm under water level. The test started by placing the mouse in the pool at different quadrants in each training and the mouse were allowed to find the platform within a maximum trial time of 90 s. The trial was completed if the mice reached the platform within that time and was allowed to remain on the platform for 10 s before returning. If the mouse was unable to find the platform within 90 s, it was guided to the platform and allowed to stay for 10 s. The training was performed on 4 successive days with two trials per day, with one hour gap between. The probe trial was performed on the fifth day with the platform removed from the pool. Initiation started from the farthest direction from the position of platform used during training. The time spent in the platform quadrant and the number of crossing the platform location were calculated. The activities of the mice in all behavioral tests were recorded using a video tracking camera and analyzed using ANY-maze software (Stoelting Co. USA).
miRNA sequencing data processingTotal RNAs from plasma samples were extracted using mirVana miRNA Isolation Kit (Cat.No.AM1561, Ambion, Austin, TX, US) following the manufacturer’s instructions. The RNA concentrations were determined by the absorbances at 260 nm using NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Libraries for sequencing were prepared using 10 ng of total RNA using QIAseq miRNA Library Kit (Cat.No. 331502, Qiagen, Germantown, MD, USA). In brief, adapters are ligated sequentially to the 3’ and 5’ ends of miRNAs, followed by universal cDNA synthesis, cDNA cleanup, library amplification and library cleanup. Constructed libraries were quantified and validated using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and 2400 bioanalyzer (Agilent Technologies, USA) system, and were then sequenced on an Illumina NovaSeq 6000 system using PE150 model (Shanghai Biotechnology Corporation) at an average read depth of 20–35 million reads per sample.
Raw reads were processed with fastx (version 0.0.13, http://hannonlab.cshl.edu/fastx_toolkit/) to remove adaptors, low-quality reads, and reads shorter than 10 nt to obtain clean reads. The clean reads (18–40 nts) were aligned to miRNAs in miRbase20.0 (http://www.mirbase.org/) using Bowtie2 software (version 2.4.4) [22, 23]. Read counts were normalized using the trimmed mean of M-values (TMM) method, and then referenced the sequencing depth to get values of transcripts per million (TPM) [24, 25]. In addition, for unannotated miRNAs, we used miRCat pipeline in the UEA sRNA workbench (version 4.4) to evaluate potential novel miRNA candidates according to miRNA precursor hairpins [26].
Bioinformatic analysismiRNAs were further filtered by expression level to exclude low expression genes using the ‘filterByExpr’ function and a count value > = 10 in over 50% of the samples in each group (n > 7 in AD group and n > 6 in control group) was used as cut-off. The gene counts were normalized by the method of trimmed mean of M-values using the ‘calcNormFactors’ function of edgeR. Principle component analysis of the normalized miRNA expression data after filtering were performed using the ‘prcomp’ function and the differentially expressed miRNAs (DEMs) between the controls and AD patients was analyzed using the ‘glmQLFit’ function of edgeR [27, 28]. The fold-change of at least 2 with adjusted p-value (FDR) < 0.05 was used as the cutoffs. The heatmap of DEMs was generated using ‘pheatmap’ package and the relation between groups was depicted by hierarchically clustered according to Pearson correlation and average linkage. The volcano plot, which the x-axis represents the log2-transformed fold change and the y-axis the log10-transformed p-value was constructed by using ‘ggplot2’ package The above process is all implemented in R (version 4.2.1). Comprehensive Automatically Mined Database of Human Diseases (Malacards) was used to search cognitive dysfunction related genes for a gene list. Online analysis tools of TargetScan (version 8.0) and microT-CDS (version 5.0) were used for miRNA target gene prediction. ‘VennDiagram’ package of R software (version 4.2.1) was employed to depict the intersections of target genes of DEMs and cognitive dysfunction related genes. The miRNA-mRNA regulatory network was visualized by using Cytoscape (version3.9.1). Functional annotation of Gene Ontology (GO, Biological Process, Cellular Component, Molecular Function) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of cognitive dysfunction related miRNA targets were performed using DAVID (v2022q2, https://david.ncifcrf.gov/) based on total miRNA targets as a population background.
miRNA mimics, recombinant AAV vectors and miR-140-miR-122 TuD expression vectorsMimics of miR-140 and miR-122, and scramble miRNA were purchased from GenePharma (Shanghai, China). The sequences of miR-140-3p and miR-122-5p were obtained from miRBase database (www.mirbase.org). The sequence of the stem loop structure of these two miRNAs (Table S4) were cloned into the expression plasmids GV412 (Genechem, Shanghai, China). The AAV vector GV412 contains CMV promoter and encoding eGFP. The 293T cells were transfected with the recombinant AAV vectors, pHelper and pAAV9 rep-cap using Lipofectamine 2000. After transfection for 48 h, cells were harvested and lysed by four freeze-thaw cycles in dry ice and 37℃ water baths for collect the virus. The lysed incubation with benzonase (Sigma Aldrich) at 37℃ for 30 min to concentrate virus. Then, the virus was purified by the CsCl gradient ultracentrifugation and were further concentrated on Amicon-15 Centrifugal Filters (Merck Millipore, Burlington, MA, USA). AAV genome copies were titrated using quantitative PCR (qPCR).
DNA sequences encoding tough decoy RNAs targeting both miR-140-3p and miR-122-5p, designed according to Haraguchi et al., were synthesized by Sangon Biotech (Shanghai, China) and were cloned into the NheI/HindIII sites of pcDNA3.1 [29]. The miR-140-miR-122-TuD contains two tandem stem-loop structures, each of which contains either the mmu-miR-140-3p or mmu-miR-122-5p binding sites flanked by an 18-bp long stem and a 26-bp stem-loop, respectively. A 4-nt long mismatch was inserted into each of the miRNA-binding sites between 10 and 11 from the 3’-end to avoid complete paring. The schematic representation and sequence of the miR-140-miR-122-TuD is shown in Fig. 2. The sequences of all constructs were verified by sequencing (Sangon Biotech, Shanghai).
Fig. 1
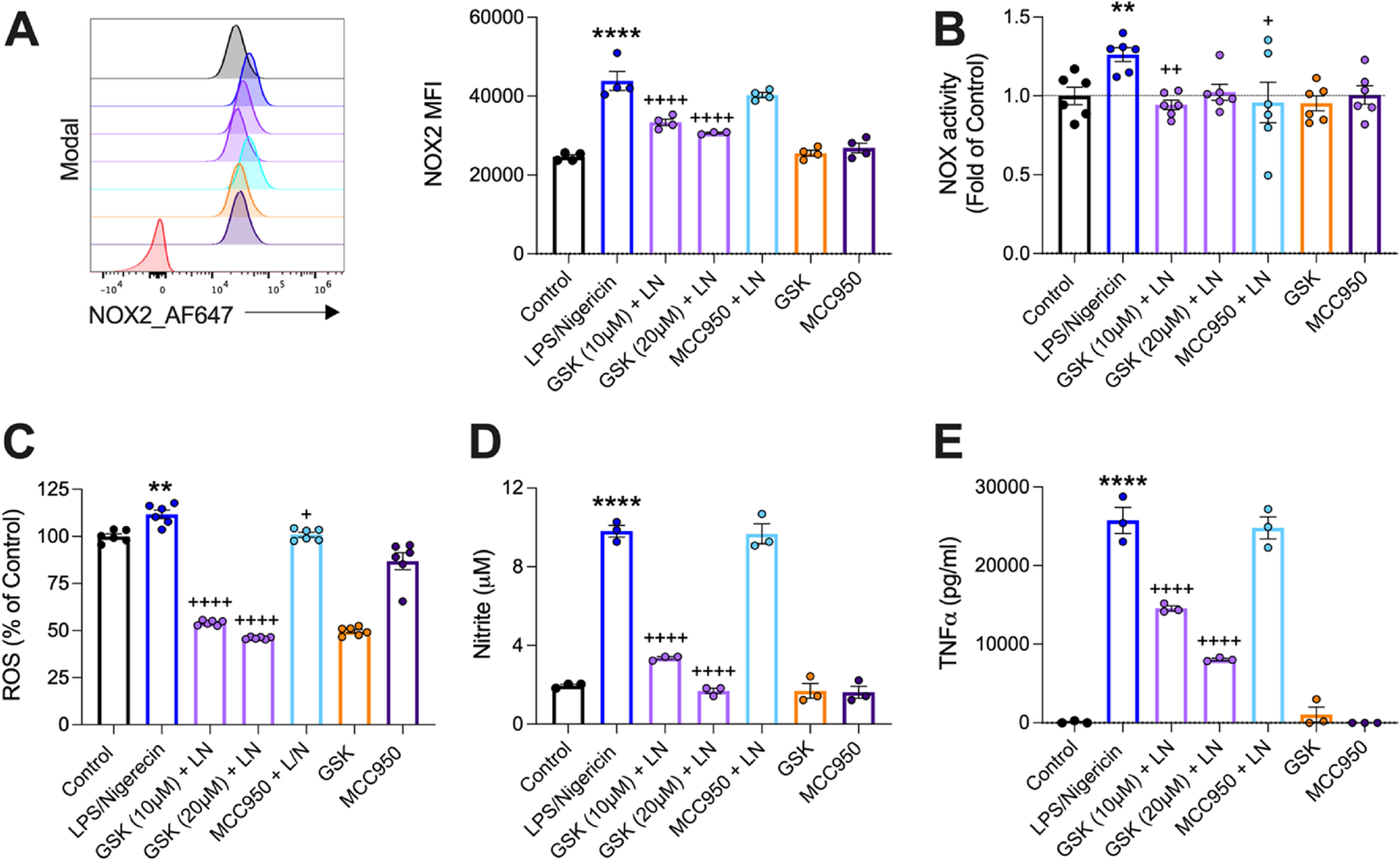
miRNA profiling of the blood plasma of AD patients and cognitively normal controls. (A). PCA analysis of the normalized expression values of plasma miRNA, showing the separation between AD patients and controls on the first principal component. (B). Heat map showing the clustering of differentially expressed miRNAs. (C). Volcano plot displaying the fold change and p value of DEMs, with the labeling of all DEMs with |log2-fold change| >1 and FDR adjusted p < 0.05. (D). miRNA–mRNA regulatory network visualization for the enrichment analysis of DEMs. The size of miRNA nodes indicated the number of their target genes. Red circles represent DEMs and pink/green circles are their target genes, which green circles also are cognitive dysfunction related genes. (E) Gene Ontology biological process terms and Kyoto Encyclopedia of Genes and Genomes pathways in enrichment analysis of DEMs targets. Dot size represents the fold enrichment. The color of each dot represents the category of ontology, and the X-axis represents the p-value. (F). Quantitative RT-PCR validation of the expression of miR-140 and miR-122 in the plasma of AD patients and controls. The results were presented as means ± SD (n ≥ 9). (G-H). Quantitative RT-PCR data showing the expression of miR-140 (G) and miR-122 (H) in the cortex and the hippocampi of C57BL/6J mice and the APP/PS1 mice, respectively. The results were presented as means ± SD (n ≥ 3). *p < 0.05, ** p < 0. 01. Statistical analyses were performed using a two-tailed unpaired Student’s t-test
Fig. 2
Dysregulated expression of miR-140 and miR-122 affected sAPPα production via targeting ADAM10. (A). The specific inhibitory sequence of miR-140-miR-122-TuD containing two tandem stem-loop structures was cloned into the NheI/HindIII sites of pcDNA3.1. (B). The sequences of miR-140-miR-122-TuD, with each of the stem -loop contains two miRNA-binding sites flanked by an 18-bp long stem and a 26-bp stem-loop, respectively. A 4-nt long mismatch was inserted into each of the miRNA-binding sites between 10 and 11 from the 3’-end to avoid complete paring. (C). Quantitative real-time PCR showing significantly upregulated or downregulated miR-140 and miR-122 expression levels in 293T cells transfected with mimics of miR-140 and miR-122 or pcDNA3.1-miR-140-miR-122-TuD. Untransfected cells and cells transfected with scramble miRNA and pcDNA3.1 were used as controls. The results were presented as means ± SD (n = 5). *p < 0.05, ****p < 0.0001. Statistical analyses were performed using One-way ANOVA. (D-I). Cultured 293T cells (D-F) and HT22 cells (G-I) were transfected with: (1) vehicle only; (2) scramble miRNA; (3) mimics of miR-140; (4) mimics of miR-122; (5) mimics of miR-140 and miR-122; (6) pcDNA3.1; (7) pcDNA3.1-miR-140-miR-122-TuD; (8) mimics of miR-140 and miR-122 + mock treatment; (9) mimics of miR-140 and miR-122 + 2 µM atRA. The control cells were treated with vehicles. (D-F). Western blot analysis and densitometric quantification of the relative expression of ADAM10 (D), sAPPα (E) and APP (F) in the transfected 293T cells as indicated. The results were presented as means ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analyses were performed using One-way ANOVA or two-tailed unpaired Student’s t-test. (G-I). Western blot analysis and densitometric quantification of the relative expression of ADAM10 (G), sAPPα (H) and APP (I) in the transfected HT22 cells as indicated. The results were presented as means ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analyses were performed using One-way ANOVA or two-tailed unpaired Student’s t-test. (J). ELISA analysis of the soluble Aβ42 monomer in the cultured HT22 cells transfected with scramble miRNA, mimics of miR-140 and miR-122, pcDNA3.1 and pcDNA3.1-miR-140-miR-122-TuD. The results were presented as means ± SD (n = 3). *p < 0.05, statistical analyses were performed using One-way ANOVA
RNA extraction and quantitative real time-PCR (qRT-PCR)For isolation of miRNAs from blood plasma, the miRNAs were isolated from 100 µL of blood plasma using the miRNeasy Serum/Plasma Kit (Cat.No. 217184, Qiagen, Germantown, MD, USA) according to the manufacturer’s protocols. 1 pmol synthetic Caenorhabditis elegans miRNA cel-miR-39 was used as an internal standard and added to each sample. The final RNA was eluted with 10 µL of nuclease-free water and 3.75 µL RNA of each sample was reverse transcribe using the Mir-X miRNA First-Strand Synthesis Kit (Cat.No. 638313, Takara, Ohtsu, Japan). The primer pairs consist of a common mRQ 3’ primer and one of the specific 5’ primers (hsa-miR-140-3p: 5’-AAGTTGCATACCACAGGGTAGA-3’, hsa-miR-122-5p: 5’-AACACGCTGGAGTGTGACAA-3’, cel-miR-39: 5’-ACCGAGGTTCACCGGGTGTAA-3’) were used to amplify hsa-miR-140-3p, hsa-miR-122-5p and cel-miR-39, respectively.
For RNA isolation from mouse tissues, the mouse hippocampal and cortical tissues were first homogenized and the total RNA was extracted using Trizol-chloroform and isopropanol. 1 µg of total RNA was reverse transcribe using specific stem-loop primer (miR-140-3p: 5’-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCCGTGG-3’, miR-122-5p: 5’-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCAAACA-3’, snRNA-U6: 5’-CGCTTCACGAATTTGCGTGTCAT-3’) to synthesize cDNAs using the PrimeScript RT reagent Kit with gDNA Eraser (Cat. No. RR047A, Takara, Kusatsu, Japan). The primer pairs (5’-AAGTTGCATACCACAGGGTAGA-3’ and 5’-CAGTGCAGGGTCCGAGGT-3’, 5’-AACACGCTGGAGTGTGACAA-3’ and 5’-CAGTGCAGGGTCCGAGGT-3’) were used to amplify mmu-miR-140-3p and mmu-miR-122-5p, respectively. The primer pairs (5’-CCGTTCCTGCGTTCTTAT-3’ and 5’- ACTGGTTGCCCTTGGTAG-3’, 5’-ATCTGACCTTCCTGCCCTCCAC-3’ and 5’-ACCCAAGAGCACACACGACATTC-3’) were used to amplify Chi3l1 and H2-Ea cDNA. cDNA of Gapdh was amplified using the primer pairs (5’-GGCAAATTCAACGGCACAGTCAAG-3’ and 5’-TCGCTCCTGGAAGATGGTGATGG-3’) and was used as the internal reference of gene expression. Quantitative real-time PCR was performed using TB Green Premix Ex TaqII (Tli RNaseH Plus) (Cat. No. RR820A, Takara, Kusatsu, Japan) on an Applied Biosystems Quanstudio3 platform (Applied Biosystems, Waltham, MA). The relative expression of genes was calculated using the 2−ΔΔCt formula and were normalized using internal controls of cel-miR-39, U6 and Gapdh, respectively.
Western blotThe cells and mouse brain tissues were lysed in RIPA buffer (Sigma) containing PMSF and phosphatase inhibitor cocktail. Protein concentration was determined using BCA Protein Assay Kit (Thermo Fisher Scientific). 20 µg proteins of each sample were separated on 10% SDS-PAGE gel and were then transferred onto PVDF membranes (BioRad). The membranes were blocked with 5% skim milk in 0.1% TBS-Tween 20 for 1.5 h under room temperature and probed with primary antibodies overnight at 4℃. The membranes were washed three times with 0.1% TBS-Tween 20 and probed with HRP labeled secondary antibodies for 2 h at RT. The immunoblots were visualized using Amersham Imager680 (GE Healthcare Bio-Sciences Corp, Piscataway, NJ) and the bands intensities were quantified by densitometric analysis with ImageJ software (ImageJ 1.8.0).
Quantification of Aβ plaques, number of microglia cells, Iba1-positive areas, microglia coverage and dystrophic neurites using immunohistochemistryMice were anesthetized with 2% pentobarbital sodium (50 mg/kg) and perfused with 0.9% saline. The mouse brain was removed in ice and fixed in 4% paraformaldehyde (PFA) overnight at 4℃, followed by graded sucrose solutions (20%, 30% in PBS) at 4℃. Hemisphere brains were embedded in O.C.T. compound and store at -80℃. 15-µm and 30-µm-thick serial coronal sections of hippocampus region were prepared with a cryostat microtome (CM1950, Leica). For immunolabeling of Aβ deposits, 15-µm-thick sections were washed with PBS and permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature (RT). After blocking with 10% goat serum in PBS at RT for 1 h, the tissue sections were subjected to overnight incubation at 4℃ with primary antibodies (MOAB-2, 1:1000, Cat. No. 13075, Novus Biologicals, Littleton, CO, USA and anti-Iba1, 1:1000, Cat. No. 016-20001, Wako Chemical, diluted in PBS + 1.5% goat serum), respectively. The sections were then washed with 0.1% Triton X-100 in PBS 5 min for 3 times, followed by incubation with Alexa Fluor 647-conjugated Affinipure F(ab′ )₂ Goat Anti-Mouse IgG (1:1000, Cat. No. 115-605-003, Jackson ImmunoResearch Laboratorie) and Cy3-AffiniPure F(ab′ )₂ fragment Goat anti-Rabbit IgG (1:1000, Cat. No. 111-166-144, Jackson ImmunoResearch Laboratorie) for 1 h at room temperature, respectively. The nuclei were counterstained using Hoechst 33342 (1:10000, Cat. No. 14533, Sigma-Aldrich) in Fluoromunt™ Aqueous Mounting Medium (Cat. No. F4680; Sigma-Aldrich) and coverslipped before imaging. The unilateral hippocampal regions of three consecutive coronal sections visualized using Olympus FV3000 confocal microscope and the images were captured at 10× magnification. The number and size of MOAB-2 positive Aβ plaques as well as Iba1-positive areas were quantitated using ImageJ.
30-µm-thick tissue sections were used for microglial enveloping analysis. The tissue sections were sequentially permeabilized with 0.5% Triton X-100 in PBS for 1 h at RT, blocked in 10% goat serum + 0.5% Triton X-100 in PBS for 1 h at RT, and probed with indicated primary antibodies (anti-Iba1, 1:100, Cat. No. sc-32,725, Santa cruz biotechnology, anti-lamp1, 1:800, Cat. No. ab24170, Abcam, diluted in 0.5% Triton X-100 containing 1.5% goat serum) overnight at 4℃. The sections were then incubated with Goat Anti-Mouse IgG (1:1000, Cat. No. 115-605-003, Jackson ImmunoResearch Laboratorie), and Cy3-AffiniPure F (ab′) ₂ fragment Goat anti-Rabbit IgG (1:1000, Cat. No. 111-166-144, Jackson ImmunoResearch Laboratorie) for 2 h at RT, respectively. Briefly washed 3X in PBS, the sections were stained with 1% Thioflavine S for 8 min at RT, followed by washing in 70% ethanol for 4 min and rinsing with water. The slides were mounted in Fluoromunt™ Aqueous Mounting Medium (Cat. No. F4680; Sigma-Aldrich) and coverslipped before imaging. The unilateral hippocampal fields of three consecutive coronal sections were visualized using Olympus FV3000 confocal microscope and the z-stacked images were captured using a 40 × objective at 1024 × 1024-pixel resolution at 3 μm z-step. The number and size of ThioS-positive Aβ plaques as well as the number of microglial cells within 25 μm radius from the center points of the Aβ plaques were quantified using ImageJ. The neuritic dystrophy associated with compact amyloid plaques was measured using ImageJ according to the method described by Condello et al., 2015 [30]. A Z-projection of three optical slices through the center of the plaque was generated for each of the plaques and was used to calculate microglia coverage as well as the dystrophic neuritic area. The colocalization of the plaques and Iba1-positive areas was recognized using ImageJ. The intersections between the colocalized areas and the plaque perimeter, which was defined using ImageJ, were identified manually for each of the plaque. The proportion of the summed arcs of the perimeter with Iba1 colocalization was defined as microglia coverage. To compare the dystrophic neurite formation in areas with or without microglia coverage, the angles used to define microglia coverage were further extended to the limit of Lamp1-positive areas (plaque area excluded), which were recognized using ImageJ. The Lamp1-positive areas within this extended selection as well as the Lamp1-positive areas outside the selection were calculated, respectively and were normalized to their respective angular degrees. The significance of correlation between microglia coverage and Lamp1-positive areas in different size of Aβ plaque was tested.
Mouse embryonic primary cortical cell culturePrimary cortical tissues were isolated from E15.5 C57BL/6J embryo [31, 32]. The pregnant female mice (E15.5) were euthanized with CO2 and the embryos were collected in ice-cold Hank’s balanced salt solution (HBSS, Invitrogen). The cortical tissues of the embryos were dissected in fresh pre-chilled HBSS under a stereomicroscope, followed by digestion in 0.25% trypsin at 37 °C for 15 min. The cortical tissues were then resuspended in plating medium (BME medium + 10% FBS + 0.45% glucose (20%) + 1% sodium pyruvate + 1% glutamine + 1% Penicillin/streptomycin) and triturated with care using a pasteur pipettes until a single-cell suspension was obtained. Dissociated cells were inoculated in six-well plates that were coated with poly-L-lysine (1 mg/mL) and were allowed for settle in an incubator filled with 5% CO2 at 37 °C for 4 h. The medium was then replaced with neuron medium (Neurobasal medium + 2% B27 + 1% glutamine + 0.5% penicillin/streptomycin). The cell purities were assessed 24 h later using immunofluorescence with Tubulin β3 a neuronal marker and > = 90% of neuronal cells was obtained.
pEGFP-N1 plasmids were then used to transfect neuronal cells alone or co-transfected with indicated oligos using Lipofectamine2000 to improve visualization of neuron morphology [33]. Half the medium was replaced with fresh medium containing either sAPPα (10 nM) or atRA (2 µM) or vehicles before processed for further analysis. Z-stacked images acquired with Olympus FV3000 confocal microscope (at 1 μm step, 40 × objective) of at least 30 randomly chosen GFP-positive cells from each sample were used for Sholl analysis to calculate the number of dendrites intersections on concentric circles at 5 μm intervals using ImageJ [34].
Cell lines and cultured cell transfectionsThe human embryonic kidney 293T cells, the mouse hippocampal neuronal cell line HT22 and mouse microglial cell BV2 were purchased from ATCC (Manassas, Virginia, USA). 293T, HT22 and BV2 cells were cultured in the Dulbecco’s modified Eagles medium (DMEM) with 10% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin at 37℃ in a humidified atmosphere with 5% CO2. 293T and HT22 cells were plated on 6-well plates at the density of 4 × 105/ well. The cells were transfected with 50 nM RNAs or 1 µg indicated plasmids using Lipofectamine 2000 transfection reagent according to the manufacturer’s instructions. The cell culture medium was replaced with fresh medium containing indicated treatments 24 h after transfection and maintained before processed to further analysis at indicated time points.
ELISAThe amount of Aβ1–42 monomer in cells and tissues were determined using Amyloid beta 42 Mouse ELISA Kit (Cat. No. KMB3441, Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. The hemisphere hippocampal tissues were lysed in RIPA buffer (8 µL/ 1 mg of tissue) for 30 min on the ice, followed by centrifugation at 12,000 × g for 30 min. 5 µL supernatant of each sample was used for Aβ42 ELISA assay. The cells in 12-well plates were homogenized in RIPA buffer (60 µL per well) for 30 min on the ice. 50 µL supernatant of each sample was used for Aβ 42 ELISA assay.
Golgi staining and dendrite arborization analysisThe FD Rapid Golgi Stain Kit (Cat. No. PK401, FD Neurotechnologies) was used for mouse brain to Golgi staining according to manufacturer’s instruction. Briefly, the mouse brain hemispheres (n = 4) obtained immediately after sacrifice were immersed into mercuric chloride solution (mixture of solutions A and B) for 2 weeks. The samples were then transferred to a cryoprotectant solution (solution C) and were kept in dark for another week. The samples were then frozen in isopentane on the dry ice and stored at -80℃. 100-µm thick serial coronal sections of hippocampus region were obtained using a cryostat microtome (CM1950, Leica). The slices were stained with ammonium hydroxide solution (mixture of solution D and E) for 10 min at room temperature, followed by 2 × brief wash with water. Slices were dehydrated with graded alcohols and cleared with xylenes, and then coverslipped using neutral balsam (BL704A, Biosharp, Hefei, China). Z-stack images (at 1 μm steps) of pyramidal neuron from hippocampus CA1 regions were captured under 20 × objective using EVOS FL Auto microscope (Thermo Fisher Scientific, Waltham, USA) and 3D images were reconstructed using Vaa3D software [35]. 10–15 pyramidal neurons per mouse were selected for Sholl analysis using ImageJ. The dendrite intersections on concentric circles with radius increased by 5 μm each starting from the cell soma were quantified.
Phagocytosis assay of FITC-conjugated Aβ1–42FITC-conjugated Aβ1–42 peptide (Cat. No. M212900, MREDA, Beijing, China) was dissolved in DMSO to make the 200 µM stock solution and stored at − 80 °C before use. 1 µM FITC-Aβ42 working solution was made 3 days before use by adding DMEM to the stock solution and incubation at 37 °C to allow for fibrillar Aβ aggregation. HT22 cells were transfected with indicated oligos or constructs for 12 h, followed by co-culturing with BV2 cells for 12 h. Mock-transfected cells were used as controls. Fibrillar Aβ solution was added to the co-cultured cells to a final concentration of 500 nM and incubated for 2 h to allow for Aβ phagocytosis. The cells were then fixed with 4% PFA at room temperature for 15 min and permeabilized with 0.2% Triton X-100 in PBS for 10 min with brief washes before and after. After blocking with 10% goat serum and 2% BSA in PBST for 1 h at room temperature, the cells were probed with anti-Iba1 (1:500, Cat. No. 019-19741, Wako, Japan) overnight at 4 °C. After washing with PBST three times, the cells were then incubated with Alexa Fluor 647 conjugate secondary antibody (1:800, Cat. No. 111-606-004, Jackson ImmunoResearch) for 1 h at room temperature. Cell nuclei were stained with Hoechst 33342 (1 µg/mL, Cat. No. B2261, Sigma-Aldrich) at room temperature for 10 min. The z-stack confocal images were captured at 0.7 μm step using 100× oil immersion objective and the 3D representative images were created using ImageJ’s 3D Viewer plugin. Nine optical fields were randomly chosen for each experiment and the z-stack images were captured under 40 × objective at 1 μm step size using Olympus FV3000 confocal microscope (Olympus, FV3000, Tokyo, Japan). The images were processed with FV31S-SW Viewer software (version 2.5) before quantification analysis of Aβ phagocytosis. The online software cellpose (version 2.0) was used for cellular segmentation, and ImageJ was used to assess the number of Aβ+ microglia and analysis the mean fluorescence intensity of FITC-Aβ1–42.
Statistical analysisAll statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software Inc., San Diego, CA, USA). Comparisons between two groups were performed using a two-tailed unpaired Student’s t-test. Multiple comparisons were performed using a one-way ANOVA followed by post hoc test. Data are expressed as mean ± SD or mean ± SEM, as indicated in the relevant figure legends. A probability of p < 0.05 was considered statistically significant.
Comments (0)