Mice
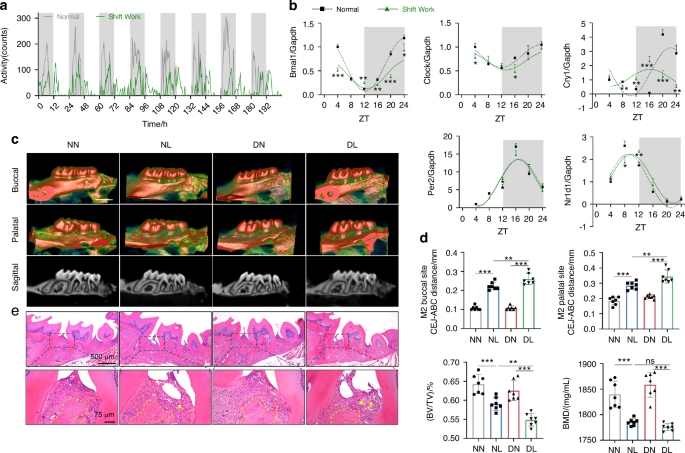
Six-week-old C57BL/6J male mice were kept under normal light-dark cycles, with lights turned on at 6 A.M. and turned off at 6 P.M. To induce circadian rhythm disruption by simulated shift work, the mice were exposed to a condition where the lights were turned on 8 h in advance every 1–2 days for 8 weeks, i.e., turning the lights on at 10 P.M. and off at 10 A.M. according to the previous methods.48,49 For voluntary running wheel assessing, mice were placed in individual cages equipped with voluntary running wheels and exposed to normal or disturbed light-dark cycles for 8 days. Activity profiles were generated by calculating the counts of running wheel spins per 30 min. To construct the model of circadian rhythm disruption complicated with periodontitis, wire was ligated around the first and second maxillary molars of mice for 14 days in the seventh week of abnormal light exposure. To inhibit pyroptosis, Z-YVAD-FMK (MedChem Express, HY-P1009, Monmouth Junction, NJ, USA) was administered into three palatal gingival sites of the experimental maxillary molars every 2 days at a concentration of 200 μmol/L (2 μL) per site using a 33-gauge needle Hamilton syringe (Hamilton Company, Reno, NV, USA) at ZT 4 (10 A.M.). For restoring the level of BMAL1, SR8278 (MedChem Express, HY-14415) was injected intraperitoneally at a concentration of 500 μmol/L (110 μL) at ZT 4. All animal studies were carried out in accordance with the committee guidelines of the Animal Care Committee of the Fourth Military Medical University (Approval No. IACUC-20230117).
Micro-CT
The maxillae were collected and fixed in 4% paraformaldehyde for 24 h and then scanned using the Quantum GX2 Micro-CT Imaging System (PerkinElmer, Waltham, MA, USA). The indicators including the bone mineral density (BMD), BV/TV, and the distance from the CEJ to the ABC were used to evaluate the degree of alveolar bone loss and tissue damage.
RNA extraction and quantitative real-time PCR (qRT-PCR)
For tissue RNA extraction, the buccal and palatal gingiva of the experimental second maxillary molars were collected. Cell samples were harvested as usual. After being washed three times with phosphate-buffered saline (PBS; Procell Life Science & Technology, Wuhan, China), the gingiva or cells were immersed in the AG RNAex Pro Reagent (Accurate Biology, AG21102, Changsha, China). Total RNA was extracted following the manufacturer’s instructions. An Evo M-MLV RT Premix for qPCR kit (Accurate Biology, AG11706) was used to prepare the cDNA synthesis reactions according to the manufacturer’s instructions. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as the reference gene. qRT-PCR was performed on the CFX96 Touch Real-Time PCR Detection System (Bio Rad, CA, USA) by using a SYBR® Green Premix Pro Taq HS qPCR kit (Accurate Biology, AG11701), and the relative expressions of target mRNAs were calculated by the 2−ΔΔCT method. The primer sequences used in this study are listed in Table S1.
Western blot
The proteins were harvested from gingiva and cells by using Cell Lysis Buffer for Western and IP (Beyotime, P0013J, Shanghai, China) with ProtLytic Protease Inhibitor Cocktail (New Cell & Molecular Biotech, P001, Suzhou, China). After separated by Tris-glycine SDS-polyacrylamide gel electrophoresis (7.5% or 10%), the proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Merck Millipore, IPVH00010, Billerica, MA, USA) and blocked with QuickBlock™ Blocking Buffer for Western Blot (Beyotime, P0252). The PVDF membranes were incubated overnight at 4 °C with antibodies against BMAL1 (ABclonal, A17334, Wuhan, China), CLOCK (ABclonal, A7265), CRY1 (ABclonal, A6890), PER2 (ABclonal, A13168), NR1D1 (ABclonal, A20452), NLRP3 (R&D Systems, MAB7578, Minneapolis, MN, USA), CASPASE1 (Proteintech, 22915-1-AP, Wuhan, China), GSDMD (Santa Cruz Biotechnology, sc-393581, Santa Cruz, CA, USA), IL-1β (ImmunoWay Biotechnology, YT5201, Plano, TX, USA), β-Actin (ImmunoWay Biotechnology, YM3029). After incubation with Peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) (Jackson, 115-035-003, West Grove, PA, USA), HRP Goat Anti-Rabbit IgG (H + L) (ImmunoWay Biotechnology, RS0002), HRP Goat Anti-Rat IgG (H + L) (ABclonal, AS028), the bands were visualized by using an enhanced chemiluminescence kit (AccuRef Scientific, AP0082S, China) and imaged by the ChemiDoc XRS+ System (Bio Rad).
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed by using NIH3T3 cell line, which was purchased from the Cell Repository of the Chinese Academy of Sciences (Shanghai, China), according to the manufacturer’s instructions of ChIP Assay Kit (Beyotime, P2078). Briefly, upon reaching 90% confluence in a 100 mm dish, cells were cross-linked for 10 min using 1% formaldehyde (Sigma-Aldrich, F8775, MO, USA). Genomic DNA was fragmented to a size of 200 to 500 bp using sonication, and immunoprecipitated with either an anti-BMAL1 antibody (Cell Signaling Technology, 14020S, MA, USA) or an IgG antibody (Proteintech, 30000-0-AP). The genomic DNA that had been detached and purified was amplified by qRT-PCR with the primers listed in Table S2.
Inclusion and exclusion criteria of clinical periodontal tissue collection
The collection of clinical samples has been approved by the Ethics Committee of the Stomatology Hospital of the Fourth Military Medical University (Approval No. IRB-REV-2022046). The healthy periodontal tissues involved in the experiment were collected from the healthy third molars or premolars, which were extracted from patients aged 18 to 35 years old, with no periodontal attachment loss, redness, and swelling caused by periodontitis. The inflammatory periodontal tissues were obtained from patients aged 18 to 35 years old who underwent flap surgery due to severe periodontitis. The clinical attachment loss of the tooth was ≥5 mm, and the X-ray showed that the alveolar bone resorption exceeded 1/2 to 2/3 of the root length, which met the diagnostic criteria for stage III to VI periodontitis. The patients of tissue origin have no systemic disease.
Histological analysis
The gingival tissues from patients and the maxillae from mice were fixed in 4% paraformaldehyde for 24 h, after which the samples were washed with PBS several times. With or without decalcified with 10% ethylenediaminetetraacetic acid for 20 days, the samples were dehydrated using either alcohol or sucrose solution for paraffin-embedded sections or cryosections, respectively. Paraffin-embedded sections were stained with standard H&E for physiopathological analysis and immunolabeled with antibodies targeted N-GSDMD (1:500, ABclonal, A22523), IL-1β (1:200, ImmunoWay Biotechnology, YT5201), IL18 (1:100, Abcam, ab243091, Cambridge, MA, USA), TNF-α (1:100, ImmunoWay Biotechnology, YT4689) for immunohistochemistry analysis. For dual-color immunofluorescence analysis, anti-VIMENTIN (1:100, ABclonal, A19607) and anti-GSDMD (1:100) were used to identify pyroptosis in the gingiva. Images were collected by Lecia Microsystems (M205FA, Leica, Germany) and Olympus laser confocal fluorescence microscopy (LCFM, FV1000, Olympus, Japan).
mRNA sequencing
The gingival total RNA was extracted using Trizol reagent (Thermo Fisher Scientific, 15596018, Waltham, MA, USA) following the manufacturer’s instructions. After purification, the mRNA was fragmented into short fragments and reverse-transcribed to cDNAs. Following adding an A-base, the fragmented cDNAs were ligated to the adapter containing a T-base overhang, and size selection was performed using AMPureXP beads. Amplified with PCR, the average insert size of the final cDNA libraries was (300 ± 50) bp. And at last, the 2 × 150 bp paired-end sequencing (PE150) was performed on an Illumina Novaseq™ 6000 (LC-Bio Technology, Hangzhou, China) following the vendor’s recommended protocol. Bioinformatic analysis was performed using the OmicStudio tools available at https://www.omicstudio.cn/tool.
Cells
Primary mGFs isolated from gingival tissues of the healthy mice, were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Procell Life Science & Technology), 100 U/mL penicillin, and 100 mg/mL streptomycin (BioCytoSci, Burlington, VT, USA) in a 37 °C incubator with 5% CO2 and 95% humidity. To simulate the inflammatory challenge, mGFs were treated with P. gingivalis LPS (InvivoGen, tlrl-pglps, San Diego, CA, USA) at a concentration of 5 μg/mL. For the transfection assay, when the mGFs reached 70% confluence in six-well plates, 75 pmol of Bmal1 siRNA (Santa Cruz Biotechnology, sc-38166) or 1 μg of Bmal1 plasmid (Gene Pharma, Shanghai, China) were transfected into mGFs using X-treme GENE HP DNA Transfection Reagent (Roche Diagnostics, 51572200, Mannheim, Germany), prior or posterior to LPS treatment. For NR1D1 inhibition, mGFs were treated with SR8278 at a concentration of 2.5 μmol/L after P. gingivalis LPS treatment.
Cytotoxicity assay
Cellular cytotoxicity was detected by PI staining and LDH activity assay. Following LPS treatment and transfection, mGFs were harvested, rinsed with PBS, and stained with 10 μg/mL of PI (Beyotime, ST511) for 20 min. Subsequently, Antifade Mounting Medium containing Hoechst 33342 (Beyotime, P0133) was used for nuclear counterstaining. Images were collected by Olympus LCFM. Meanwhile, the supernatants of mGFs were centrifuged and transferred to fresh EP tubes to measure the LDH activity using CheKine™ Micro LDH Assay Kit (Abbkine Scientific, KTB1110, Atlanta, GA, USA) according to the manufacturer’s instructions. The absorbance was detected at 450 nm with a microplate spectrophotometer (BioTek, Epoch, Winooski, VT, USA).
SEM detection
The mGFs were rinsed with PBS five times and fixed in 2.5% glutaraldehyde (Leagene, DF0156, Beijing, China) at 4 °C overnight, after which the cells were washed in PBS several times and dehydrated with graded ethanol. Following critical point drying by hexamethyldisilazane, the samples were sputtered with gold and observed using an FE-SEM (Hitachi S-4800, Japan).
Dual-luciferase reporter assay
Wild-type (WT) mouse Gsdmd promoter and its mutant (MUT) deleting the E-box motif (5’-ACACGTGGGA-3’) were cloned into a GPL4-Basic luciferase reporter construct (Gene Pharma), respectively. L929 cells were transfected with the GPL4-Basic plasmid (empty vector), GPL4-Luci plasmid (control vector expressing firefly luciferase), the indicated reporter plasmids, GPL4-RL plasmid (control vector expressing Renilla luciferase), and either Bmal1 plasmid or empty vector using Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA). After transfection for 48 h, cells were harvested and the luciferase activities were measured using a Dual-Luciferase Reporter Gene Assay Kit (Beyotime, RG027) by using a VICTOR Nivo Multimode plate reader (PerkinElmer, HH35000500, Finland).
Statistical analysis
The data are displayed as the mean ± SD of at least three independent experiments. Data analysis was performed using GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA). Unpaired Student’s t-tests were used to assess the significant differences between two independent groups, while one-way or two-way ANOVA multiple comparisons were used to compare multiple independent groups. Differences were considered significant at the values of *P < 0.05, **P < 0.01, and ***P < 0.001.




Comments (0)