
Remember me




This prospective randomized controlled trial was conducted at Zonguldak Bülent Ecevit University Hospital between January and March 2024. Ethical approval was obtained from the Local Ethics Committee (protocol number: 2023-12-13/12; ClinicalTrials.gov identifier: NCT06225908). All patients received detailed information about the study, and both written and verbal informed consent were obtained. Participants were educated on the use of the Numerical Rating Scale (NRS), the Quality of Recovery (QoR)-15 questionnaire, and the Patient-Controlled Analgesia (PCA) device. The study was conducted in adherence to the Consolidated Standards of Reporting Trials guidelines.
Study populationSixty patients, aged 18–65 years, with an American Society of Anesthesiologists Physical Status (ASA PS) I-III, scheduled for elective unilateral oncoplastic breast surgery (modified radical mastectomy and axillary lymph node dissection) under general anesthesia, were included in the study. Exclusion criteria included patients who declined participation, had ASA PS IV–V, a known allergy to local anesthetics, coagulopathy, infection at the block site, those who will not undergo axillary dissection, chronic analgesic use, were uncooperative, or were undergoing bilateral surgery. Patients were randomized into two groups using a computer-assisted program and the sealed envelope method: SPSIPB group (n = 30) and control group (n = 30). Randomization and preparation of sealed envelopes were performed by different non-study staff.
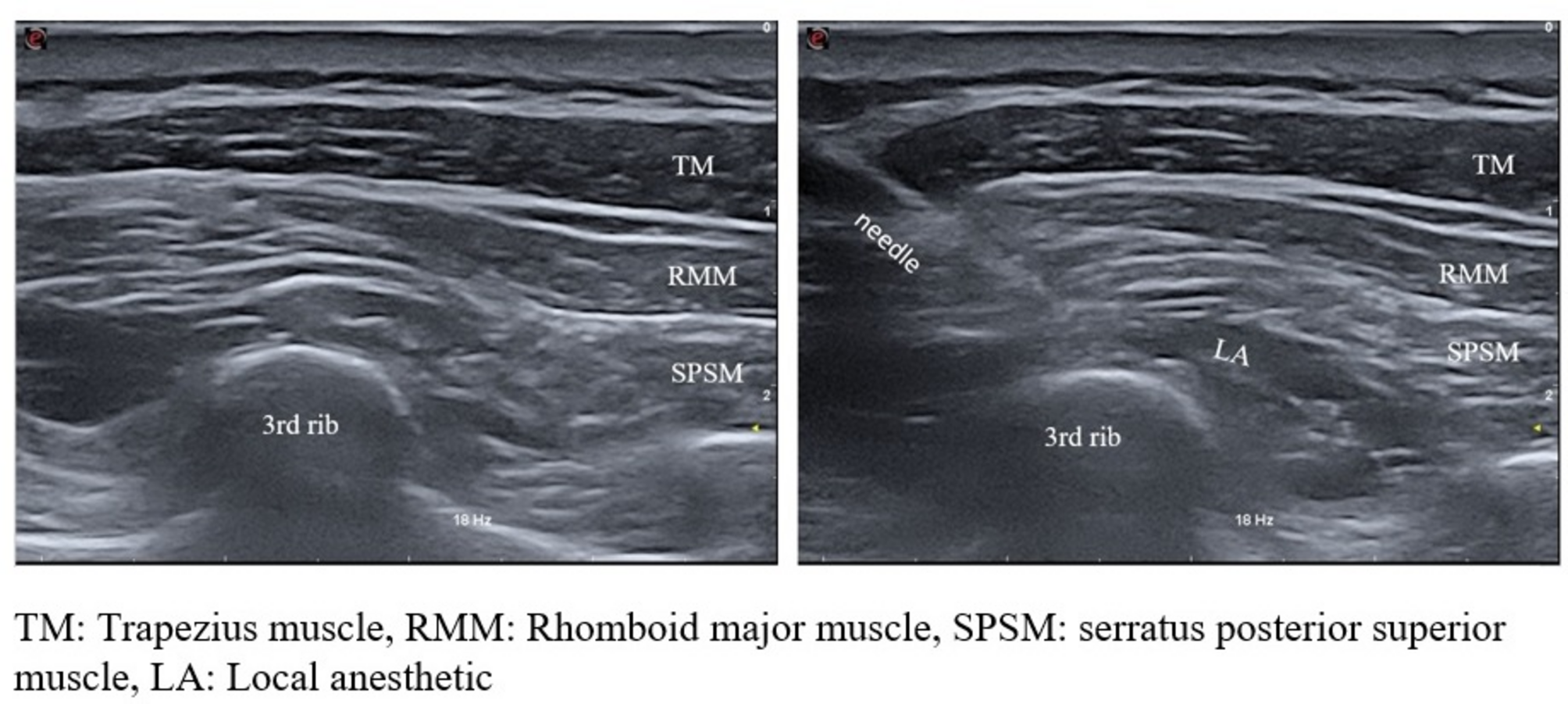
Serratus posterior superior intercostal plane blockTo ensure standardization of the study, all blocks were administered by the same anesthesiologist (ÇB), who had previously performed this block at least 50 times. Patients in the SPSIPB group were transferred to a block room before surgery. Following routine monitoring, 1 mg of midazolam was administered for sedation. Patients were positioned prone, and after ensuring proper sterilization, the SPSIPB procedure was performed using a linear ultrasound probe (3–13 mHz, MyLab X7; Esaote, Genoa, Italy). The probe was positioned along the medial border of the scapula at the level of the second and third ribs on the surgical side. Key anatomical structures, including the trapezius, rhomboid major muscle (RMM), and serratus posterior superior muscle (SPSM), as well as the second and third ribs, were identified. Using the in-plane technique, the needle was advanced into the fascial plane between the third rib and the SPSM. Proper placement was confirmed by injecting 1–2 mL of isotonic solution, followed by the administration of 20 mL of 0.25% bupivacaine after negative aspiration (Fig. 1). Twenty minutes after the block procedure, patients were transferred to the operating room.
Fig. 1
Ultrasound image of SPSIPB application
General anesthesiaGeneral anesthesia was administered to all patients, with routine monitoring supplemented by bispectral index monitoring. Intubation was performed following standard induction using intravenous lidocaine (1 mg/kg), propofol (2–3 mg/kg), fentanyl (1 µg/kg), and rocuronium (0.6 mg/kg). All surgeries were performed by the same surgeon. During the procedure, bispectral index values were maintained between 40 and 60 using sevoflurane, and remifentanil was infused at a rate of 0.05–1 µg/kg/min, as needed. Hemodynamic data were recorded at specific intervals throughout the intraoperative period. Intravenous paracetamol (10 mg/kg), tramadol (1 mg/kg), and metoclopramide (10 mg) were administered 30 min before the end of the operation. Remifentanil infusion was discontinued with the final suture, and the total amount consumed was recorded. The time from the final suture to extubation was recorded and defined as the emergence time. At the conclusion of surgery, all patients were extubated using appropriate doses of neostigmine and atropine, and subsequently transferred to the post-anesthesia care unit (PACU). The durations of anesthesia and surgery were recorded.
Postoperative analgesia protocolPatients were monitored in the PACU, where a PCA device containing tramadol was provided. The PCA settings included no basal infusion, a 10 mg bolus dose, and a 20-minute lockout interval. The time of arrival at the PACU was designated as hour 0. Resting and dynamic (coughing) NRS scores were assessed in the PACU. For patients with an NRS score of ≥ 4, 25 µg of intravenous fentanyl was administered as a rescue analgesic. Pain was reassessed 15 min later, and if the NRS score remained ≥ 4, an additional dose of fentanyl was given. The total amount of rescue analgesics administered before discharge from the PACU was recorded. Any complications, including nausea, vomiting, hypotension, bradycardia, respiratory depression, or pruritus, were documented during the PACU stay. Patients were transferred to the ward once a Modified Aldrete score of ≥ 9 was achieved.
In the ward, 10 mg/kg of paracetamol was administered every 8 h throughout the postoperative period. Resting and dynamic NRS scores were evaluated and recorded at 1, 2, 6, 12, and 24 h postoperatively. During these evaluations, the Ramsey Sedation Scale scores and any complications were also documented. For uncontrolled pain (NRS ≥ 4) despite the use of PCA and paracetamol, 75 mg of intramuscular diclofenac sodium was given as a rescue analgesic. If the pain persisted following intramuscular injection, 0.25 mg/kg of intravenous pethidine was administered. The amount of rescue analgesics administered postoperatively was recorded. Treatments for nausea and vomiting were documented over the first 24 h postoperatively. At the 24-hour mark, patients completed the QoR-15 questionnaire again. The PCA device was then terminated, and the total opioid consumption and the number of PCA requests over 24 h were recorded.
The primary outcome was the cumulative opioid consumption within the first 24 h postoperatively. Secondary outcomes included resting and dynamic NRS pain scores, QoR-15 questionnaire scores, intraoperative remifentanil consumption, and the incidence of postoperative nausea and vomiting.
Sample sizeThe sample size was estimated using the G*Power program (Heinrich Heine University, Düsseldorf, Germany) based on a pilot study conducted prior to the main study. For the pilot, 10 patients not included in the final analysis were enrolled in each group. The calculation was based on the total opioid consumption within 24 h postoperatively, using a 95% confidence interval, and a power of 90% (d = 0.9). The opioid consumption was recorded as 44 ± 35.96 mg in the SPSIPB group and 96.2 ± 73.22 mg in the control group. Based on these parameters, the minimum required sample size per group was determined to be 28. To account for potential dropouts, 32 patients were included in each group.
Statistical analysisStatistical analyses were conducted using IBM SPSS software (version 20.0; IBM Corp., Armonk, NY, USA). The Kolmogorov–Smirnov test was applied to assess the normality of data distribution. Continuous data are presented as mean ± standard deviation. Categorical variables were analyzed using the chi-square test. For normally distributed data, the Student’s t-test was employed, whereas the Mann–Whitney U test was used for data that did not follow a normal distribution. Statistical significance was set at p < 0.05. Bonferroni correction was used for the analysis of NRS. Statistical significance was adjusted to p < 0.0083 due to measurements from six time points.
Comments (0)