
Remember me
Our research complies with all relevant ethical regulations including the International Society for Stem Cell Research Guidelines for Stem Cell Research and Clinical Applications of Stem Cells. Experiments with human pluripotent stem cells and blastoids were approved by the Scientific Ethics Committee for Hovedstaden (H-21043866/94634). HNES1 line use is with agreement of the Steering Committee of the UK Stem Cell Bank (SCSC23-30). The WiCell line H9 (WA09) was used under the agreement 23-W0460. Extended blastoid culture was approved by the Scientific Ethics Committee for Hovedstaden (H-21043866/94634 and H-24048289). This work did not exceed a developmental stage normally associated with 14 consecutive days in culture after fertilization nor did it entail any implantation in vivo. Human embryos or gametes were not used in this study.
Cell cultureExperiments were performed with the human embryonic stem cell lines H9 (WA09, WiCell) and HNES1 (Nichols Lab)41. Both cell lines were provided by the laboratory of J. Brickman. H9 cell line was chemically reset according to ref. 12. Cell lines were regularly tested for mycoplasma and were negative.
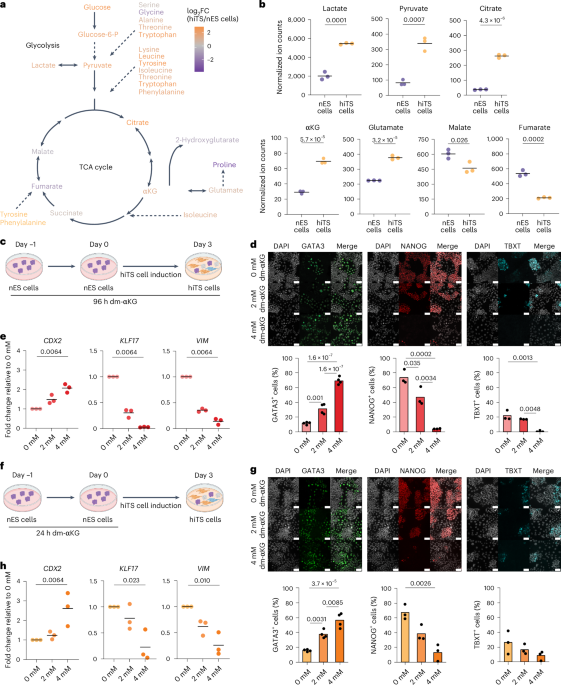
Human naive ES cell cultureHuman nES cells were cultured on gelatin-coated plates including a feeder layer of gamma-irradiated DR4 mouse embryonic fibroblasts (ATCC, SCRC-1045) in tt2iLGö medium or PXGL medium. tt2iLGö medium consists of N2B27 basal medium supplemented with PD0325901 (1 μM, Axon MedChem, Axon 1408), CHIR99021 (0.3 μM, Axon MedChem, Axon 1386), Gö 6983 (2 μM, Axon MedChem, Axon 2466) and human leukaemia inhibitory factor (hLIF, 10 ng ml−1, Qkine, Qk036)12. PXGL medium consists of N2B27 basal medium supplemented with PD0325901 (1 μM, Axon MedChem, Axon 1408), XAV939 (2 μM, Merck Sigma-Aldrich, X3004), Gö 6983 (2 μM, Axon MedChem, Axon 2466) and hLIF (10 ng ml−1, Qkine, Qk036)54. N2B27 basal medium contains DMEM–F12 with GlutaMAX (50%, Thermo Fisher Scientific, 31331028), neurobasal medium (50%, Fisher Scientific, 11570556), N2 supplement (1×, Merck Sigma-Aldrich, SCM012), B27 supplement (0.5×, Thermo Fisher Scientific, 17504044), 2-mercaptoethanol (0.2 mM, Thermo Fisher Scientific, 31350010) and GlutaMAX (0.75 mM, Thermo Fisher Scientific, 25030024). Cells were cultured in hypoxic conditions (5% CO2, 5% O2) and passaged as single cells every 3–4 days. Cells were cultured for 24 h with Y27632 (10 μM, Axon MedChem, Axon 1683) after passaging. Medium was changed daily. Treatment of nES cells with dm-αKG, A-485 or UNC1999 was performed by directly diluting dimethyl 2-oxoglutarate (2 mM or 4 mM, Merck Sigma-Aldrich, 349631), A-485 (1 μM or 5 μM, Selleckchem, S8740) or UNC1999 (1 μM, BioNordika, CST-46080S) in the culture medium before medium change. Cell numbers were determined using the Via1-Cassette in the NucleoCounter NC-200 (Chemometec).
hiTS cell inductionhiTS cell induction was performed according to Dong et al., with minor modifications20. In brief, before the induction, nES cells were depleted from the feeder layer by incubating the single-cell suspension on 0.1% gelatin for 45–75 min. Human nES cells then were seeded at a density of 1.8 × 104 cells cm−2 in a 6-well plate precoated with ECMatrix-511 Silk E8 Laminin Substrate (Merck Sigma-Aldrich, CC161) in tt2iLGö medium. After 24 h, the medium was changed to hiTS cell medium consisting of N2B27 supplemented with bovine serum albumin (BSA) (0.3%, Sigma, A3294), heat-inactivated foetal bovine serum (0.2%, Thermo Fisher Scientific, 12389782), insulin-transferrin-selenium-ethanolamine (ITS-X, 1×, Gibco, 51500), l-ascorbic acid (1.5 μg ml−1, Sigma, A8960-5G), recombinant human EGF (50 ng ml−1, Qkine, Qk011), CHIR99021 (2 μM, Axon MedChem, Axon 1386), A83-01 (0.5 μM, Axon MedChem, Axon 1421), SB431542 (1 μM, Axon MedChem, Axon 1661), valproic acid (0.8 mM, Axon MedChem, Axon 3127) and Y27632 (10 μM, Axon MedChem, Axon 1683). Cells were cultured in hypoxic conditions (5% CO2, 5% O2), and passaged as single cells or small clumps every 3–4 days. The medium was changed daily. Treatment of hiTS cell induction with dm-αKG, A-485 or UNC1999 was performed by directly diluting dimethyl 2-oxoglutarate (2 mM or 4 mM, Merck Sigma-Aldrich, 349631), A-485 (1 μM or 5 μM, Selleckchem, S8740) or UNC1999 (1 μM, BioNordika, CST-46080S) in the culture medium before medium change.
nEnd inductionnEnd induction was performed according to ref. 16 with minor modifications. In brief, before the induction, nES cells were depleted from the feeder layer by incubating the single-cell suspension on 0.1% gelatin for at least 45 min. Human nES cells then were seeded at a density of 2.2 × 104 cells cm−2 in a 6-well plate precoated with ECMatrix-511 Silk E8 Laminin Substrate (Merck Sigma-Aldrich, CC161) in tt2iLGö medium. After 24 h, the medium was changed to RACL medium consisting of RPMI 1640 with GlutaMAX (Gibco, 61870044) with B27 minus insulin (Gibco, A1895601) supplemented with activin A (100 ng ml−1, Qkine, Qk005), CHIR99021 (3 μM) and hLIF (10 ng ml−1). Treatment of the nEnd induction with dm-αKG was performed by directly diluting dimethyl 2-oxoglutarate (2 mM or 4 mM, Merck Sigma-Aldrich, 349631) in the culture medium before medium change.
Blastoid formationBlastoid experiments were performed according to Kagawa et al., with minor modifications21. In brief, nES cells cultured in PXGL were collected by incubation for 3 min with Accutase (Merck Sigma-Aldrich, A6964) and were depleted from the feeder layer by incubation on 0.1% gelatin for at least 60 min. Single cells were plated at a density of 80–85 cells per microwell of a 24-well AggreWell 400 plate (Stemcell Technologies, 34415) in N2B27 supplemented with BSA (0.3%, Sigma, A3294) and Y27632 (10 μM, Axon MedChem, Axon 1683) to aggregate for 16 h. Subsequently, half medium was changed with 2× PALLY consisting of N2B27 supplemented with BSA (0.3%, Sigma, A3294), PD0325901 (1 μM, Axon MedChem, Axon 1408), A83-01 (1 μM, Axon MedChem, Axon 1421), hLIF (10 ng ml−1, Qkine, Qk036), oleoyl-l-α-lysophosphatidic acid sodium salt (LPA, 1 μM, Merck Sigma-Aldrich, L7260) and Y27632 (10 μM, Axon MedChem, Axon 1683). Twenty-four hours later, half medium was changed with 1× PALLY. After 48 h of PALLY culture, blastoids were maintained for 56 h in LY medium consisting of N2B27 supplemented with BSA (0.3%, Sigma, A3294), LPA (1 μM, Merck Sigma-Aldrich, L7260) and Y27632 (10 μM, Axon MedChem, Axon 1683). Treatment of blastoid induction with dm-αKG was performed by directly diluting dimethyl 2-oxoglutarate (4 mM, Merck Sigma-Aldrich, 349631) in the culture medium. The medium was then equilibrated for 2–3 h at 37 °C in hypoxic conditions (5% CO2, 5% O2) before use.
Blastoid attachment assaysAfter 120 h blastoid induction, blastoids treated with 0 mM or 4 mM dm-αKG for 40 h were selected with a P10 pipette and transferred either on 4-well IbiTreat μ-plates (Ibidi, 80426) in IVC1 medium or on an ECM mix in 8-well IbiTreat μ-plates (Ibidi, 80826) in post-implantation medium. First, the IVC attachments were performed according to Shahbazi et al. with minor modifications56. In short, blastoids were cultured directly in IVC1 medium for 24 h before assessing attachment. After 48 h in IVC1, the medium was changed to IVC2 medium. IVC1 medium consists of advanced DMEM–F12 (Thermo Fisher Scientific, 11540446) supplemented with heat-inactivated foetal bovine serum (20%, Thermo Fisher Scientific, 12389782), GlutaMAX (2 mM, Thermo Fisher Scientific, 25030024), ITS-X (1×, Gibco, 51500), β-oestradiol (8 nM, Merck Sigma-Aldrich, E2758), progesterone (200 ng ml−1, Merck Sigma-Aldrich, P0130) and N-acetyl-l-cysteine (25 μM, Merck Sigma-Aldrich, A7250). IVC2 medium is composed of advanced DMEM–F12 (Thermo Fisher Scientific, 11540446) supplemented with knockout serum replacement (30%, Gibco, 10828028), GlutaMAX (2 mM, Thermo Fisher Scientific, 25030024), ITS-X (1×, Gibco, 51500), β-oestradiol (8 nM, Merck Sigma-Aldrich, E2758), progesterone (200 ng ml−1, Merck Sigma-Aldrich, P0130) and N-acetyl-l-cysteine (25 μM, Merck Sigma-Aldrich, A7250). Attachment efficiency was quantified for 7 biological replicates consisting of 80 structures each (N = 7, n = 560). Attached structures were collected 4 days after attachment.
The attachments on the ECM mix were performed according to Karvas et al. with guided modifications57. In brief, an 8-well IbiTreat μ-plate (Ibidi, 80826) was coated with an ECM mix consisting of Cultrex UltimaMatrix (80%, R&D System, BME001-05) and hES-cell-qualified Matrigel (20%, Corning, 11573560) for at least 1 h at 37 °C. Blastoids were transferred on the solidified ECM mix in post-implantation medium. Post-implantation medium or N2B27 + E2 consists of DMEM–F12 with GlutaMAX (50%, Thermo Fisher Scientific, 31331028), neurobasal medium (50%, Fisher Scientific, 11570556), N2 supplement (0.5×, Merck Sigma-Aldrich, SCM012), B27 supplement (0.5×, Thermo Fisher Scientific, 17504044), 2-mercaptoethanol (0.2 mM, Thermo Fisher Scientific, 31350010), GlutaMAX (2 mM, Thermo Fisher Scientific, 25030024), non-essential amino acids solution (1×, Gibco, 11140050), sodium pyruvate (1 mM, Gibco, 11360070) and β-oestradiol (10 nM, Merck Sigma-Aldrich, E2758). Medium was changed daily. At days 4 and 5, the size of the structures was measured (diameter, µm) (N = 4, n = 142). Structures were collected 7 days after attachment.
Real-time quantitative PCRRNA was collected and extracted using the Qiagen RNeasy Mini Kit (Qiagen, 74104) with on-column DNAse treatment. Subsequently, cDNA was produced using Superscript III Reverse Transcriptase (Invitrogen, 18080093). RT-qPCR was performed using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25778) in a LightCycler480 (Roche). Quantification was performed by applying the comparative cycle threshold (Ct) method. Relative expression levels were normalized to TBP. The primers used during RT-qPCR analysis are summarized in Supplementary Table 5.
mRNA-seqRNA samples were prepared as for RT-qPCR. Samples collected included nES cells treated with dimethyl sulfoxide (control, n = 4) or 4 mM dm-αKG (n = 2) or 1 μM A-485 (n = 2) for 24 h, and day 3 hiTS cells derived from these cells collected at day 3 of induction. Libraries for sequencing were prepared using 500 ng of total RNA and the NEBNext Poly(A) mRNA and NEBNext UltraII stranded RNA-seq library preparation kit. Ten cycles of amplification were performed. Samples were sequenced using NextSeq2000 P2 Reagents (100 cycles) v3 (Illumina) with paired-end 61-bp sequencing.
Raw sequencing data were demultiplexed and converted into FASTQ files using Illumina’s bcl2fastq (v2.20.0.422; RRID: SCR_015058), assigning an average of 31 million read pairs per library. Quality check was done with FastQC (v0.11.9; RRID: SCR_014583). Paired-end reads were aligned with STAR (v 2.7.2d; RRID: SCR_004463 (ref. 65)) to the reference genome GRCh38.p13 with gene annotations from GENCODE release 32 (RRID: SCR_014966 (ref. 66)) and option ‘--quantMode GeneCounts’ was set to count the number of reads per gene while mapping.
Differential gene expression analysis was performed using DESeq2 (1.36.0; RRID: SCR_015687 (ref. 67)) in R (v 4.2.1). Genes with fewer than ten raw read counts across samples were removed, and size factors were estimated for each sample. For nES cells and day 3 hiTS cells separately, DESeq was run with ‘design = ~treatment’, and results were extracted for dm-αKG versus control and P300i (A-485) versus control.
For GSEA, results for the P300i treatment were ranked by Wald statistics, and genes significantly up- or downregulated upon dm-αKG treatment (adjusted P value <0.05) were defined as separate gene sets. The R package fgsea (v 1.22.0; RRID: SCR_020938 (ref. 68)) was used to perform preranked GSEA with parameters ‘nPermSimple = 10000, eps = 0’, and defaults otherwise.
IF microscopyCells were cultured in Ibidi microwell plates and fixed with 2% paraformaldehyde for 10 min at room temperature. After fixation, paraformaldehyde solution was removed, and the samples were washed at least three times with 1× phosphate-buffered saline (PBS; Gibco, 14190144). The samples were then permeabilized for 20 min using 0.2% Triton X-100 (Merck Sigma-Aldrich, T8787) and afterwards blocked using blocking buffer containing 1% BSA (Merck Sigma-Aldrich, A7979), 0.1% Tween20 (Merck Sigma-Aldrich, P1379) and 10% normal donkey serum (Merck Sigma-Aldrich, D9663) in 1× PBS for at least 3 h. The samples were then incubated overnight at 4 °C with primary antibodies diluted in blocking buffer. The next day, samples were washed with 1× PBS containing 0.1% Tween20 (PBST) at least three times for 10 min each. After washing, the samples were incubated with secondary antibodies diluted in blocking buffer for 3 h in the dark at room temperature. The samples were then washed with PBST three times for 10 min each and afterwards mounted using Vectashield mounting medium (VWR, VECTH-1000) or 1× PBS. The antibodies used for IF assays are listed in Supplementary Table 5.
For blastoid and aggregate IF, the same protocol was followed with minor modifications. Namely, blastoid samples were fixed with 4% paraformaldehyde for 20 min at room temperature. Subsequently, the samples were washed for three times 10 min with PBST supplemented with BSA (0.3%). For image quantification of aggregates, sum projection of five consecutive confocal z-stacks was used. These stacks were 3 μm apart and at a central location of the aggregate. Nuclei were segmented in ilastik and curated in ImageJ using the DAPI signal. aPKCζ signal was used to identify the whole region of interest of each aggregate. For aPKCζ polarity quantification, the area between the end of the outer nuclei and the cell membrane was quantified and assigned as the apical signal. For each aggregate, the average apical intensity was divided by the remaining non-nuclear signal. For YAP and TAZ quantifications, signals were measured for each nucleus and categorized on the basis of their location either within the most outer layer or inner nuclei. Shown is the average IF intensity normalized to the outer cell signal.
After attachment, blastoid IF was performed following a similar protocol. In brief, the samples were fixed with 4% paraformaldehyde for 3 h at 4 °C, while gradually removing ECM. After fixation, paraformaldehyde solution was removed, and the samples were washed at least three times with 1× PBS. The samples were then permeabilized for 20 min using 0.3% Triton X-100 with 0.001% polyvinyl alcohol (PVA) (Merck Sigma-Aldrich, 363170) and afterwards blocked using blocking buffer containing 0.001% PVA, 1% BSA, 0.1% Tween20 and 10% normal donkey serum in 1× PBS for at least 5 h. The samples were then incubated for 48 h at 4 °C with primary antibodies diluted in blocking buffer. Subsequently, samples were washed with 1× PBS containing 0.1% Tween20 and 0.001% PVA (PBST) at least three times for 30 min each. After washing, the samples were incubated overnight with secondary antibodies diluted in blocking buffer in the dark at 4 °C. The samples were then washed with PBST three times for 30 min each and afterwards mounted using Vectashield mounting medium (VWR, VECTH-1000).
For H3K27ac/phospho-histone H3-Ser10 staining, IF was performed on aggregate cryosections. In this case, after fixation of the aggregates (see above), they were washed three times with PBST, transferred to 15% sucrose–PBS for 10 min at room temperature and then embedded in optimal cutting temperature compound and frozen on dry ice. Frozen blocks were stored at −80 °C and then sectioned at 8-µm thickness with a cryostat (Leica). Sections were placed on Superfrost Plus slides (ThermoFisher), air dried and stored at −80 °C.
For staining, slides were defrosted and air dried for 10 min. Sections were permeabilized for 15 min with 0.5% Triton X-100–PBST, rinsed three times with PBST and incubated for 1.5 h in blocking buffer. After blocking, primary antibodies in blocking buffer were added for 3.5 h, at room temperature. Next, samples were washed three times with PBST, followed by a 1-h incubation with secondary antibodies in blocking buffer. Finally, the sections were washed three times with PBST, which was supplemented with DAPI on the second wash (5-min incubation). Sections were mounted with SlowFade Diamond Antifade Mountant (Invitrogen) and a coverslip and subsequently sealed with nail varnish.
Image quantification of H3K27ac in cryosections was performed similar to YAP and TAZ in whole aggregates, with minor modifications. Here, the sum projection of three consecutive confocal z-stacks (z-size 2 µm) was used. After nuclear segmentation, outer versus inner cells of the aggregate were manually selected in each image and the fluorescence intensity of DAPI and H3K27ac was measured. Background signal was subtracted from the values, and H3K27ac intensity was normalized to DAPI. Shown is the average H3K27ac intensity in outer versus inner cells.
Gas chromatography–mass spectrometry time-of-flight (GC–MS-TOF) analysisCell samples were washed three times with 1× PBS before adding 1.0 ml ice-cold 90% methanol with labelled internal standards (Supplementary Table 5). The samples were collected by scraping the surface of the wells with a cell scraper and stored in −80 °C. Samples were thawed on ice with subsequent snap-freezing in liquid nitrogen, thawing on ice and vortexing for 10 s, which was repeated three times. Next, the samples were incubated on ice for 1 h, to facilitate the precipitation of proteins. Sample tubes were centrifuged at 16,000g or high speed for 15 min at 4 °C. Fifty microlitres of the resulting supernatant was transferred to gas chromatography vials and were dried at room temperature using a nitrogen evaporator. The dried metabolic extracts were stored at −20 °C until further analysis. The remaining pellet was used to measure protein concentration. Briefly, the pellets were resuspended in RIPA buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate and Sigma-Aldrich protease inhibitor Cocktail cOmplete). The lysates were incubated for 10 min on ice followed by sonication (4 °C, 8 cycles, each with 30-s pulses followed by 30 s rest in between) using a Bioruptor Plus. The lysates were centrifuged for 10 min at 20,000g at 4 °C. After centrifugation, the supernatants were used to measure protein content with the DC protein assay reagent (Bio-Rad, 5000111).
GC–MS-TOF analysis was performed with a Leco Pegasus BT GC/TOFMS instrument (Leco). Before the sample analysis, an automatic two-step derivatization reaction using a Gerstel MSP multisampler. In the first step, 12.5 µl of methoxamine reagent (2% in pyridine) was added to the dried extracts, and the mixture was incubated at 45 °C for 60 min. In the second step of the reaction, 12.5 µl of N,O-bis(trimethylsilyl)trifluoroacetamide with 1% trimethylchlorosilane was added to the same mixture, which was subsequently incubated under the same reaction conditions as before. Finally, to control for injection precision, 50 µl of hexane containing 10 mg l−1 of 4,4′-dibromooctafluorobiphenyl was added to the samples. The resulting silylated metabolites were separated on a Restek Rxi 5-ms column (pn: 13423-6850) with a helium flow of 1.2 ml min−1 and an inlet temperature of 270 °C. The temperature gradient started at 40 °C, where it was kept steady for 1 min. Next, temperature was increased at a pace of 20 °C min−1 until reaching 340 °C, where it was maintained for 3 min. Ions were generated by a −70 V electron beam at an ionization current of 2.0 mA, and 10 spectra s−1 were recorded in the mass range 50–750 m/z.
For data processing, raw files were converted into centroid mode and subsequently exported as netCDF files. The in-house Swedish Metabolome Centre (www.swedishmetabolomicscentre.se) GC–MS software was used for data extraction following a targeted approach. An in-house library, based on mass spectra and retention times of specific compounds, was used to obtain a target list of metabolites and their respective peak areas. The peak areas of target compounds were normalized to internal standards. For metabolite quantification, normalized peak areas were compared with a dilution series of standards. In addition, the normalized to internal compounds peak areas were further normalized to protein concentrations.
Methoxamine reagent and N,O-bis(trimethylsilyl)trifluoroacetamide with 1% trimethylchlorosilane were purchased from Thermo Scientific. Hexane, l-valine-d8, l-glutamic acid-13C515N, succinic acid-2,2,3,3-d4 and 4,4′-dibromooctafluorobiphenyl were obtained from Sigma-Aldrich. Malic acid-13C4, citric acid-1,5,6-carboxyl-13C3, dl-2-hydroxyglutaric acid-13C5 and α-ketoglutaric acid-d6 were from Cambridge Isotope Laboratories. Standards aimed for quantification (d-glucose, d-fructose-6-phosphate, d-glucose-6-phosphate, l-lactic acid, pyruvic acid, citric acid, α-ketoglutaric acid, dl-α-hydroxyglutaric acid, succinic acid, dl-malic acid, fumaric acid, l-glutamine, cis-4-hydroxy-d-proline, d-ornithine, urea, l-tryptophan, l-alanine, l-glutamic acid, l-aspartic acid, l-proline, l-lysine, l-methionine, l-phenylalanine, l-serine, l-threonine, l-tyrosine and l-valine) were purchased from Sigma-Aldrich.
Liquid chromatography–quadrupole-time-of-flight mass spectrometry (LC–QTOF-MS) analysis of Acetyl-CoACell samples were washed three times with 1× PBS before adding 0.5 ml ice-cold 90% methanol containing 0.5 µM acetyl-CoA-13C2 as internal standard. Then, 200 μl of the resulting supernatant was transferred to liquid chromatography vials and dried at room temperature using a nitrogen evaporator. The dried metabolic extracts were stored at −80 °C until further analysis.
LC–QTOF-MS analysis was performed with an Agilent 1290 Infinity II liquid chromatograph (Agilent) connected to a Bruker Tims TOF Pro-2 mass spectrometer (Bruker Daltonics). Metabolites were separated on an Atlantis Premier BEH-Z-HILIC 1.7 µm VanGuard Fit, 2.1 × 100 mm (Waters). The mobile phases were A: 10 mM NH4 acetate with 0.05 µM medronic acid), B: 90% acetonitrile with 10 mM NH4 acetate. The flow rate was 0.35 ml min−1. The gradient started with 90% of mobile phase B, and thereafter B was decreased to 60% in 5 min and further decreased to 30% in 2 min. After being held at 30% B, the column was reequilibrated back to 90% B. The column temperature was 40 °C, and acquisition was made with a vacuum-insulated probe-heated electrospray ionization source operated in positive ionization mode. The scan speed was 2 Hz, mass range 50–1,000 m/z and mass resolution about 50,000.
The peak area of acetyl-CoA was normalized to the internal standard. For metabolite quantification, normalized peak areas were compared with a dilution series of standards. In addition, the normalized to internal standard peak areas were further normalized to protein concentrations. Data processing was performed with Bruker Compass DataAnalysis version 6.1 software and TASQ 2023b (Bruker).
Histone posttranslational modification analysisSamples were collected by 10 min of trypsinization. Subsequently, the samples were washed three times with 1 ml 1× PBS. After removing any remaining PBS, the samples were snap frozen. Samples were shipped to EpiQMAx and analysed as follows. Acid-extracted histones were processed according to a SP3 protocol as described previously69. However, proprietary steps developed by the EpiQMAx GmbH have been added to adjust the protocol for histone-specific aspects. Upon overnight digestion at 37 °C and 2,000g in a table-top thermomixer, samples were acidified by adding 5 µl of 5% trifluoroacetic acid (TFA) and quickly vortexed. Beads were immobilized on a magnetic rack, and peptides were recovered by transferring the supernatant to new PCR tubes. Samples were dried down using a vacuum concentrator and reconstituted by adding 12 µl 0.1% fluoroacetic acid (FA) to reach a peptide concentration of approximately 0.2 µg µl−1. mass spectrometry injection-ready samples were stored at −20 °C.
Approximately 200 ng of peptides from each sample were separated on a C18 column (bioZen 2.6 µm Peptide Polar-C18 150 ×0.075 mm Phenomenex) with a gradient from 5% B to 30% B (solvent A 0.1% FA in water, solvent B 80% acetonitrile (ACN), 0.1% FA in water) over 35 min at a flow rate of 300 nl min−1 (Vanquish Neo UHPLC-Systems, Thermo Fisher) and directly sprayed into a Exploris 240 mass spectrometer (Thermo Fisher Scientific). The mass spectrometer was operated in full-scan mode to identify and quantify specific fragment ions of N-terminal peptides of human histone 3.1 and histone 4 proteins. Survey full-scan mass spectrometry spectra (from m/z 250 to 1,600) were acquired with resolution 60,000 at m/z 400 (automatic gain control target of 3 × 106). The mass spectrometric conditions were as follows: spray voltage, 1.8 kV; no sheath and auxiliary gas flow; heated capillary temperature, 300 °C.
Data analysis was performed with Skyline (version 20.2.0.343)70 by using doubly and triply charged peptide masses for extracted ion chromatograms. Peaks were selected manually. Heavy arginine-labelled SpikeTides (13C6; 15N4) were used to confirm the correct retention times and for signal normalization purposes, because all heavy standards were incorporated across all samples at the same concentration. Integrated peak values (total area MS1) were used for further calculations. Endogenous posttranslational modification (PTM) signals were normalized according to the variation of the signals of the spiked-in heavy standards. The percentage of each modification within the same peptide is derived from the ratio of this structural modified peptide to the sum of all isotopically similar peptides. Therefore, the total area MS1 value was used to calculate the relative abundance of an observed modified peptide as a percentage of the overall peptide. The unmodified peptide of histone 3.1 (amino acids 41–49) was used as indicator for total histone 3.1. Coeluting isobaric modifications were quantified using three unique MS2 fragment ions. Averaged integrals of these ions were used to calculate their respective contribution to the isobaric MS1 peak (for example, H3K36me3 and H3K27me2K36me1). To calculate the incorporation of precursor M2 [U-(13) C] from glucose into histone peptides, we first calculated the normal isotope distribution for a singly acetylated peptide in the control group. The incorporation of a heavy acetyl group shows a mass shift of 2 Da in the modified peptide. We took the intensity from precursor M [U-(12) C] and divided it by the intensity from precursor M2 [U-(13) C]. Further, the incorporation of precursor M2 [U-(13) C] in the treated samples was calculated by taking the precursor M2 [U-(13) C] intensity and subtracted by the ratio between precursor M [U-(12) C] from the treated group and divided by the normal isotope distribution ratio. This calculation was used to evaluate only the singly acetylated peptides. The relative percentage of the acetylated peptide generated by de novo acetylation was evaluated using the formula: (M2 [U-(13) C]/M2 [U-(13) C] + M [U-(12) C]) × 100.
ATAC-seqATAC-seq was performed as in ref. 71. In brief, nES cells were depleted from the feeder layer by incubating the single-cell suspension on 0.1% gelatin for 45–75 min. Cells then were seeded at a density of 1.8 × 103 cells cm−2 in a 6-well plate precoated with ECMatrix-511 Silk E8 Laminin Substrate (Merck Sigma-Aldrich, CC161) in tt2iLGö medium supplemented with 0 or 4 mM dm-αKG. Twenty-four hours later, cells were trypsinized and resuspended in N2B27. After washing in PBS, 50,000 cells were lysed for 3 min on ice in lysis buffer (0.1% NP-40, 0.1% Tween20, 0.01% digitonin, 10 mM Tris–HCl pH 7.4, 10 mM NaCl and 3 mM MgCl). Lysis was stopped with ATAC-wash buffer (0.1% Tween20, 10 mM Tris–HCl pH 7.4, 10 mM NaCl and 3 mM MgCl). After pelleting the cells were resuspended in transposition mix (1× TD buffer (Illumina, cat. no. 20034197), 0.01% digitonin, 0.1% Tween20 in PBS, and TDE1 TD enzyme (Illumina, cat. no. 20034197)) and incubated for 30 min at 37 °C in a thermomixer set on 2,000g. Transposition was terminated with the DNA Binding Buffer DNA Clean and Concentrator-5 Kit (Zymo Research, cat. no. D4014), and DNA was purified. Libraries were prepared using barcoding adapters and the NebNext High-Fidelity 2x Master Mix (NEB, cat. no. M0541S). The cycling conditions used were as follows: 72 °C 5 min; 98 °C 30 s; 11× (98 °C 30 s, 63 °C 30 s); 72 °C 1 min. Libraries were purified twice using DNA Clean and Concentrator kit. Samples were sequenced using NextSeq2000 P2 Reagents (100 Cycles) v3 (Illumina) with paired-end 61-bp sequencing.
Raw sequencing data were demultiplexed and converted into FASTQ files using Illumina’s bcl2fastq (v2.20.0.422; RRID:SCR_015058), assigning an average of 38 million read pairs per library. These were further processed using the ENCODE ATAC-seq pipeline (v 2.2.2; https://github.com/ENCODE-DCC/atac-seq-pipeline; with ‘atac.genome_tsv’: https://storage.googleapis.com/encode-pipeline-genome-data/genome_tsv/v4/hg38.tsv, ‘atac.paired_end’: true, ‘atac.auto_detect_adapter’: true, and otherwise default parameters). One instance of the pipeline was run per treatment (control and dm-αKG treated), combining three replicates each. In brief, reads were trimmed and aligned to the reference genome with Bowtie 2 (ref. 72) and alignments filtered. Peak calling was performed by MACS2 (ref. 73) on each replicate, and reproducibility is assessed using the irreproducible discovery rate (IDR) framework.
Output from the two pipeline runs was merged as in the MoTrPAC ATAC-seq pipeline (https://github.com/MoTrPAC/motrpac-atac-seq-pipeline). In brief, optimal IDR peaks (idr.optimal_peak.narrowPeak.gz) from both runs were concatenated, peaks were truncated to 200 bp around summit, sorted and merged (that is, combining overlapping or ‘book-ended’ peaks using ‘bedtools merge’), and finally this ‘master peak file’ was intersected with tagalign files using bedtools coverage (bedtools v2.28.0; RRID:SCR_006646 (ref. 74)) to get counts for each replicate. Those peaks were further annotated using HOMER’s annotatePeaks.pl (v 4.11; with hg38 and default parameters75).
The count table was loaded into R (v 4.2.1), and differentially accessible peaks between dm-αKG treated and control were identified using DESeq2 (1.36.0) with default parameters. Peaks with an adjusted P value <0.01 and log2 fold change >1 were classified as more accessible in dm-αKG-treated, and likewise less accessible with adjusted P value <0.01 and log2 fold change <−1. Either peak set (with 793 and 2,479 peaks, respectively) was then checked for overrepresented motifs compared with the background set of all peaks using HOMER’s findMotifsGenome.pl (with parameters hg38 -size 200 -mask). Genomic Regions Enrichment of Annotations Tool (GREAT) analysis was performed using the R package (v 2.1.1276) for either peak set with background set of all peaks and parameters gene_sets = ‘msigdb:C2:CP’, tss_source = ‘TxDb.Hsapiens.UCSC.hg38.knownGene’.
scRNA-seqFour 2D samples were collected by 10 min of trypsinization and dissociated into single cells. Single-cavity blastoids were collected by mouth pipetting. Then, 40-h aggregates and 120-h blastoids were dissociated into a single-cell suspension by incubation with a mixture of TrypLE 10× (1×, Thermo Fisher Scientific, A1217701) and Accutase (Merck Sigma Aldrich, A6964). Each sample was incubated with 0.5 μg of unique hashtag antibody for 20 min on ice. TotalSeq hashtag antibodies (A0251-A0256, BioLegend) were used to multiplex the samples77. All samples were sorted on a BD FACSymphony S6 (BD Bioscences) resulting in a total cell recovery of around 8,000 cells for the first experiment, 8,000 cells for the second experiment and 16,000 cells for the third experiment, which were then loaded onto a Chromium Next GEM chip (10x Genomics). Further steps of library preparation were performed according to the Chromium Next GEM Single Cell 3′ v3.1 user guide with the addition of the hashtag library for demultiplexing. Combined libraries were sequenced using the NextSeq2000 P2-100 kit (Illumina) with paired-end sequencing.
Initial processing of scRNA-seq data was performed using Cell Ranger (v 6.1.2; 10x Genomics). Dual-indexed RNA and single-indexed hashtag oligo (HTO) libraries were processed in separate instances of cellranger mkfastq, using Illumina’s bcl2fastq (v2.20.0.422). FASTQ files were aligned to the human reference genome (GRCh38, v 2020-A as provided by 10x Genomics) with cellranger count (--expect-cells = 8,000, 9,500 and 9,500), calling 8,316, 9,431 and 15,133 cells, respectively. Resulting filtered feature-barcode matrices were loaded into Seurat (v 4.3.0 (ref. 78)), excluding features that were detected in fewer than three cells. Next, RNA data were normalized with default LogNormalize method, and the HTO assay was normalized with centred log-ratio transformation. Seurat’s HTODemux function was used to assign single cells back to their sample origins, resulting in 7,001, 6,012 and 10,400 cells classified as singlets. Based on quality control plots, cells with more than 15% mitochondrial counts or less than 7,000 unique molecular identifiers (nCount_RNA; this threshold was set to 5,000 for the third experiment) were removed, retaining the following number of cells per sample: 1,158 nES cells, 900 nES cells + dm-αKG, 1,577 hiTS cells, 1,711 hiTS cells +dm-αKG (from sequencing run 1), 1,201 aggregates, 1,054 aggregates + dm-αKG, 426 blastoids, 1,092 blastoids + dm-αKG (from sequencing run 2), 3,900 blastoids, and 4,974 blastoids + dm-αKG (from sequencing run 3). These data from experiment 1 were subjected to standard Seurat processing using mostly default parameters unless indicated: FindVariableFeatures, ScaleData, RunPCA, RunUMAP (dims = 1:20), FindNeighbors (dims = 1:20) and FindClusters (resolution = 0.5), resulting in eight clusters. Aggregate samples (with and without dm-αKG) from experiment 2 were processed separately, blastoid samples were merged with those from experiment 3 (using merge(merge.data=TRUE)). To both, the aggregate subset and the merged blastoids, Seurat’s CellCycleScoring function was applied using the in-built lists of S and G2M phase markers (cc.genes.updated.2019). These were subsequently regressed out using ScaleData (vars.to.regress = c(‘S.Score’, ‘G2M.Score’), features = rownames(seur)), followed by RunPCA, RunUMAP (dims = 1:20) and FindNeighbors (dims = 1:20). FindClusters was run with a default resolution of 0.8 for the aggregates and with a resolution of 0.5 for the blastoids, both resulting in seven clusters. pTE and mTE were selected on the basis of ref. 10. For pTE markers, the top 25 genes of the NR2F2 expression module were selected if they were upregulated in NR2F2-positive TE compared with NR2F2-negative (NR2F2, SLC38A1, CCKBR, SP6, FYB1, VGLL1, CYP11A1, TINAGL1, TENM3, LGALS3, RHOBTB1, MRGPRX1, CBLB, CCR7, KCNN4, GREM2, MUC15, CYP19A1, S1PR2, PLEKHF1, PGF, LCMT1-AS2, MPP1, PWWP2B and GRAMD2B). For mTE markers, the top 25 genes correlating in expression with CDX2/S100A13/16/ATP6V0A4 were selected (CDX2, RAB25, ATP6V0A4, FABP3, PRSS8, ATP6V1B1, GPRC5A, S100A13, LRP2, S100A6, ENPEP, FUOM, TAGLN2, GALNT10, SLC12A3, TMPRSS2, S100A16, TAX1BP3, PCYT2, LARGE2, ALPP, PTGES, CD53, DHCR24 and PROM1). On the subset of TE-like cells (as indicated in Fig. 5e), the expression of both mature pTE and mTE markers was scored using AddModuleScore; the difference of these is indicated as TE maturity (polar–mural signature).
To explore transcriptional differences in experiment 1 between Seurat clusters C1 (mainly nES cells) and C2 (mainly nES cells + dm-αKG), SeuratWrappers’ function RunPresto was called with parameters group.by = ‘seurat_clusters’, ident.1 = ‘C2’, ident.2 = ‘C1’, logfc.threshold = -Inf, min.pct = -Inf. This is a Presto-based implementation of FindMarkers that runs fast Wilcoxon rank-sum test and area under the receiver operating characteristics curve analysis. For preranked GSEA, results were filtered to genes that are expressed in more than 5% of cells in either C1 or C2 and ranked by AUC-0.5. Reactome gene sets were retrieved using msigdbr (v 7.5.1) with category = ‘C2’, subcategory = ‘CP:REACTOME’. fgsea (v 1.22.0) was run with parameters minSize = 15, maxSize = 500, gseaParam = 0 (that is, ‘classic’, not ‘weighted’ scoring scheme). For aggregates, markers were identified between treated and untreated cells using RunPresto with grouping by samples, and GSEA was performed in the same way, but on Hallmark gene sets, retrieved using msigdbr(category = ‘H’).
The human embryo reference was established utilized fastMNN from Batchelor (v.1.6.2)79, through the integration of previously published datasets, encompassing six human embryonic datasets spanning from early-stage in vitro cultured human blastocysts9,10,22,46 to 3D in vitro cultured human blastocysts45, and up to CS7 human gastrula44, as described in ref. 43. The pseudotime of the reference was calculated as described in ref. 43. In detail, the pseudotimes of the reference were inferred using Slingshot (v.2.6.0)80 based on the 2D UMAP embeddings of the reference. The raw cell aggregation, projection onto the human embryo reference, cell identity prediction and pseudotime inferring, were performed using the Early Embryogenesis Prediction tool43. In brief, raw cell counts were aggregated within neighbourhood nodes, computed using ‘makeNhoods’ with from miloR(v1.2.0)81, resulting in 471, 329 and 813 representative neighbourhoods for the different experiments. The aggregated counts matrix was then normalized, and corrected by mutual nearest neighbour (MNN) identification to remove batch effects, followed by transformation to the reference UMAP by using the umap_transform function from the R package uwot (v.0.1.14)82. Cell identities were predicted by an SVM classifier generated on 20 UMAP-transformed latent spaces, and the pseudotime of query cells was determined using k-nearest neighbour clustering (k = 5) with reference cells belonging to the same lineages calculated in the top 50 PCA-corrected latent spaces.
In addition, for experiment 1, the developmental stage of the representative neighbourhoods was inferred by a k-nearest neighbours algorithm (with k = 3, as implemented in the Rfast package (v 2.0.7)) based on the Euclidean distance in the 2D UMAP embedding. These labels were propagated to all cells contributing to the neighbourhood. As there is not a 1:1 mapping between cells and neighbourhoods, final labels were assigned as follows. If >50% of labels for one cell agree, this is used as the final label; if two labels are exactly 50% each, both are used (for example, ‘E6/E7’); all other cases are considered ‘ambiguous’; if a cell is not included in any neighbourhood, it is ‘unassigned’.
MicroscopyBrightfield images were acquired with a Leica DM IL LED microscope. IF images were made with the Deltavision Widefield screening microscope, Zeiss 880 airyscan, Leica Stellaris and Leica SP8 confocal microscope. All images were analysed using Fiji (ImageJ) and/or Ilastik83.
Statistics and reproducibilityscRNA-seq for the 120 h blastoid samples was performed in two independent experiments. scRNA-seq for hiTS and nES cells and 40 h aggregates were performed once. ATAC-seq, mRNA-seq, GC–MS-TOF and histone PTM quantification were each performed once, with three, two, three and three biological replicates, respectively. All other experiments in this study were repeated three times, unless stated otherwise in the figure legends. Statistics were performed using GraphPad Prism 10.4.1 (627) or R (v 4.2.1; RRID: SCR_001905). Normality and equal variances were tested where necessary. No statistical method was used to determine sample size. For the three scRNA-seq runs, 1,315, 3,419 and 4,733 droplets were excluded because they could not be assigned to one sample by Seurat’s HTODemux. In addition, 1,655, 2,239 and 1,526 cells were excluded during quality control for having more than 15% mitochondrial counts or less than 7,000, 7,000 or 5,000 unique molecular identifiers, respectively. No other data were excluded. The experiments were not randomized. The investigators were not blinded to allocation during the experiments and outcome assessment.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)