
Remember me


Four hundred fifty-four patients from two eligible HCC cohorts (TCGA-LIHC, GSE76427) were enrolled in our research for further analysis. The result of the consensus clustering algorithm revealed that k = 2 is a desirable choice for dividing the whole cohort into subtype A (n = 330) and subtype B (n = 124) (Fig. 1a, b). The K-M curve revealed that patients in subtype A had a more prolonged os than patients in subtype B (Fig. 1c). PCA analysis showed that there were considerable differences in the expression level of FRGs between the two subtypes (Fig. 1d). Patients in subtype B were younger and later stage than those in subtype A (Fig. 1e). We found most immune cells with significant differences in A and B subtypes (Fig. 1f). The infiltration of Eosinophil, Neutrophil was significantly higher in patients of subtype A compared to those of subtype B, while resting activated CD4 T cell, activated dendritic cell, immature B cell, and MDSCs had lower infiltration in patients of subtype A. Meanwhile, the analysis of critical immune checkpoints revealed high expression of PD-L1 in subtype B (Fig. 1g).
Fig. 1
FRG subtypes and clinical characteristics, tumor immune cell microenvironments of two distinct subtypes of samples. a Consensus among clusters for each category number k. b Consensus matrix heat-map defining two clusters (k = 2). c Comparison of OS among two subtypes. d PCA analysis of transcriptomes in the two subtypes. e Comparison of clinical information between the fibrosis-related clusters. f Distribution of 22 types of immune cells among these two molecular subtypes. g Expression levels of PD-L1 in the two subtypes
3.2 Construction of the prognostic modelFirstly, we identified 783 DEGs using the “limma” R package and explored the underlying biological behavior using functional enrichment analysis. This result of enrichment revealed that biosynthetic and catabolic processes and response to xenobiotic stimulus could be major biological processes (Fig. 2a). KEGG analysis indicated enrichment of drug metabolism (Fig. 2b), suggesting that fibrosis played a crucial role in drug sensitivity. We utilized the “caret” R package to randomly divide all HCC patients into a training group (n = 227) and a testing group (n = 227). Subsequently, the Lasso Cox regression was performed for 363 prognostic DEGs to obtain the optimum prognostic model (Fig. 2c). Seven genes remained with the minimum partial likelihood of deviance, of which five were risk factors. Two were favorable factors (Fig. 2e). Then, we conducted multivariate Cox regression analysis to obtain three model genes (KPNA2, LPCAT1, AKR1D1). Multiple data sets verified the expression level of the three genes in normal and tumor tissue (Figs. 2f, S2a–c). The K-M curve showed that risk genes were related to poorer prognosis and favorable genes with better prognosis (Fig. 2g). Immunohistochemical (IHC) staining protein level obtained from the HPA database showed the results of protein expression were consistent with transcription levels (Fig. 2h). We constructed the risk model based on the formula: Risk score = (0.46609* expression of KPNA2) + (0.28024* expression of LPCAT1) + (− 0.13089* expression of AKR1D1).
Fig. 2
Development of a Novel Prognostic Signature. a The GO analysis. b The KEGG analysis. c Coefficients of DEGs shown by lambda parameter. d Partial likelihood deviance versus log (lambda) drawn by LASSO algorithm and cross-validation. e Interactions in HCC. Green and purple represent favorable and risk genes, respectively. f Expression differences of model genes. g The K-M curve of model genes. h Immunohistochemical staining protein level from the HPA database
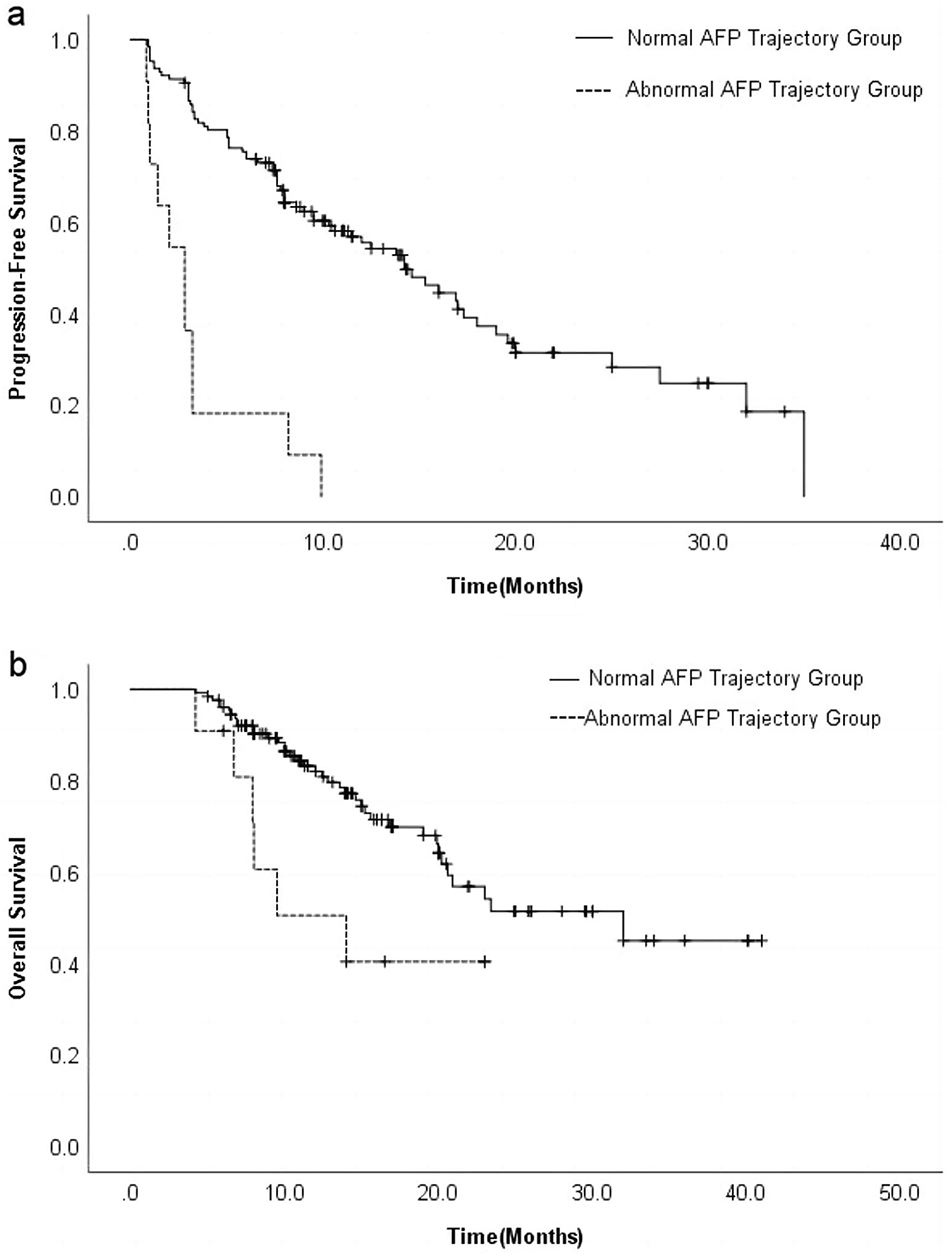
3.3 Prognostic value of model in hepatocellular carcinomaThe K-M analysis showed that high-risk HCC patients in training (Fig. 3a), testing (Fig. 3d), and all (Fig. 3g) groups had significantly lower OS compared with low-risk patients. Furthermore, the AUC of 1-, 3-, and 5-year OS was 0.749, 0.731, 0.741 in the training group (Fig. 3b), 0.695, 0.700, 0.656 in the test group (Fig. 3e), 0.721, 0.714, 0.689 in all group (Fig. 3h). In addition, we examined the model gene expression, survival status, and the distribution of HCC patients’ risk score (Fig. 3c, f, i, l). The DFS of the high-risk patients were significantly lower in train, test, and all (Fig. S3a, c, e). PCA showed discernible dimensions between high-risk and low-risk patients (Fig. S3b, d, f). In addition, patients in the ICGC set were further divided into low and high-risk groups. Similarly, we found that the OS of high-risk patients was worse than that of low-risk patients (Fig. 3j), and 1-, 3-, 5- year AUC of OS were 0.727, 0.725, and 0.753 (Fig. 3k).
Fig. 3
The overall survival analysis. a–d The K-M curve analysis in training, testing, all, validating cohort. e–h The AUC values of OS at 1-, 3- and 5- year. i–l The expression heatmap, the distribution of risk score and survival time
3.4 The relationship between risk score and clinical characteristicsThe correlation heatmap among this signature and clinical information was presented in Fig. 4a. Stage III–IV and Males were significantly related with higher risk scores compared with stage I–II and females (P < 0.05, Fig. 4b). The results of K-M curves demonstrated that our signature could be a relatively stable predictive tool for prognosis of HCC patients stratified by age (<= 65 or > 65), stage (I/II or III/IV) (Fig. 4c). To determine whether risk score could be an independent factor to predict OS of HCC patients, we performed univariate and multivariate analyses by combining risk score with the clinical characteristics. This result revealed that risk score could become a factor of independence for predicting prognosis in training, testing, all, and ICGC cohort (Fig. S4a–h). Finally, Fig. S5a–d demonstrated that the risk score could distinguish normal tissue and Cirrhosis tissue.
Fig. 4
The relationship between risk score and clinical characteristics. a The heatmap among this signature, clinical features and risk score in TCGA and GEO. b Associations of the risk score and clinical characters in TCGA and GEO. c K-M curves of the effect of clinical features on the prognosis of HCC in TCGA and GEO. d The heatmap in ICGC. e The association in ICGC. f K-M curves in ICGC
3.5 Potential pathways for the roles in HCCAs shown in Fig. S6a, DNA replication, cell cycle, mismatch repair, RNA degradation, and spliceosome were significantly enriched in the high-risk patients in the GEO and TCGA cohorts. In addition to the above-mentioned pathways, significantly enriched pathways for the ICGC cohort also included the regulation of base excision repair and nucleotide excision repair (Fig. S6b).
3.6 Construction of a nomogram to predict OSA nomogram was constructed by incorporating risk score and clinical parameters to predict the 1-, 3-, and 5- OS rates (Fig. 5a). The predictors included gender, age, stage, and risk score. Then, the calibration curve revealed that the nomogram could have a better value in predicting the prognosis (Fig. 5b). The AUC of the nomogram model revealed satisfactory accuracy for 1-, 3-, 5-year OS in training (0.787, 0.805, 0.850) (Fig. 5c), testing (0.712, 0.649, 0.640) (Fig. 5d), all set (0.749, 0.734, 0.753) (Fig. 5e), and external validation (0.787, 0.805, 0.850) sets (Fig. 5f).
Fig. 5
Construction of a 1-, 3-, 5-year OS nomogram. a Nomogram for predicting the in the all set. b Calibration curves of the nomogram for predicting OS. c–f ROC curves in the all, training, testing, ICGC sets
3.7 Relationship of risk score with immunocytes and N6-methyladenosineAs shown in Fig. 6a, the risk score was positively correlated with M2 macrophages, activated memory CD4 + T cells, and negatively correlated with M1 macrophages, naive B cells, resting mast cells, and resting memory CD4 + T cells. Further research revealed that three were significant associations between three genes and some immune cells (Fig. 6b). This result of 21 immune checkpoints showed that most immune checkpoint genes had a higher expression in high-risk patients (Fig. 6c). In addition, we analyzed the activation and inhibition of immune-related pathway in different risk score patients (Fig. 6d). Patients with high scores exhibited a significantly higher score of APC co inhibition, MHC class I and lower scores of Cytolytic activity, Type I IFN Response, Type II IFN Response compared with those with a low score. Finally, most N6-methyladenosine-related genes significantly differ in subtypes A and B (Fig. S7a), high- and low-risk sets (Fig. S7b).
Fig. 6
The evaluation of the immunocytes and N6-methyladenosine. a Correlations between risk score and immune. b Correlations between immune cells and three genes. c, d Expression of immune checkpoints and immune related pathways in the high and low-risk groups
3.8 The differential analysis of mutation, CSC index, and drug sensitivity among various risk populationsThe top 10 mutated genes were TP53, CTNNB1, TTN, MUC16, ALB, PCLO, APOB, RYR2, MUC4, FLG (Fig. 7a, b). The frequency of P53, CTNNB1, TTN, and MUC16 mutations in patients with high-risk scores was significantly higher. However, the levels of ALB and PCLO mutations were reversed. Our study of the mutation data revealed that the high-score group tends to have a higher TMB (Fig. 7c), which might account for the poorer prognosis of high-risk patients. We combined the CSC index values and risk score to evaluate the underlying relationship. We found that risk score was positively related to the CSC index (R = 0.27), suggesting that HCC cells with higher risk scores had more of a stem cell profile (Fig. 7d). Next, the chemotherapeutic agents currently used to treat HCC to assess the sensitivity in different risk groups to these agents (Fig. 7e). This results revealed that the IC50 of sorafenib, gemcitabine, docetaxel, paclitaxel, methotrexate, doxorubicin were lower in the high-risk group. At the same time, the semi-inhibitory concentrations of gefitinib, lapatinib, and other chemotherapy drugs were lower in the low-risk group.
Fig. 7
Comprehensive analysis in HCC. a The waterfall plot of somatic mutation features established of high-risk and low-risk patients. c TMB in different risk-score groups. d Relationships between risk score and CSC index. e Relationships between risk score and drug sensitivity
3.9 Testing in clinical tissue samplesIn addition to verification in the public database, we verified the accuracy of this risk model in the cohort from Qingdao Municipal Hospital. We obtained the score of each patient based on the formula. The 14 patients were divided into high-risk (n = 7) or low-risk groups (n = 7) according to the median risk score. The clinical information and risk scores of these 14 HCC patients were listed in Table 3 and visualized by heat map (Fig. 8a). Then, we analyzed the connection between the clinicopathological characters and risk score. This finding showed that The HCC patients with late-stage, high grade or liver fibrosis had a higher risk score (Fig. 8b). Finally, the results of immunohistochemistry of ki67 and H&E staining of pat.8 (the lowest risk score) and pat.5 (the highest risk score) are exhibited in Fig. 8c and d.
Table 3 The clinical information and risk scores of these 14 HCC patients from Qingdao Municipal HospitalFig. 8
Verification of the signature in our cohort. a The heatmap among clinical features and risk score. b Associations of the risk score and clinical characters (Stage, Grade, Fibrosis). c, d The immunohistochemistry of Ki67 and HE staining in HCC tissues in patient with lowest risk score and highest risk score
Comments (0)