Study Design
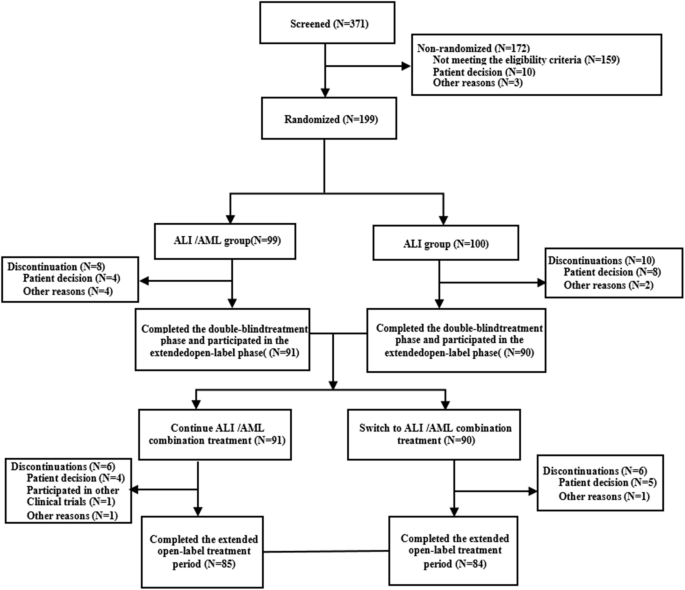
This Phase III, 52-week, multicenter, randomised, double-blind, parallel-controlled trial was conducted across 42 sites in China from January 2021 to February 2023. Patients with mild-to-moderate essential hypertension were enrolled if they were either treatment-naïve, had previously received other antihypertensive agents for ≥ 2 weeks, or had irregular treatment with ALI, and they were initially treated with ALI (240 mg/day) for a 4-week run-in period. Those with uncontrolled blood pressure (msSBP/msDBP≥140 mmHg/90 mmHg) subsequently entered a 12-week double-blind treatment period. Participants on a stable ALI regimen (240 mg/day) for ≥4 weeks with uncontrolled BP were directly randomised into the 12-week double-blind period. Following the double-blind treatment period, all participants entered the open-label treatment period, receiving the ALI/AML combination treatment for a 52-week assessment of long-term safety and tolerability.
Randomisation was performed using a stratified block design (block size = 4) with the randomisation list generated using SAS (version 9.4). Eligible patients were randomised in a 1:1 ratio, stratified by baseline office SBP (<160 or ≥160 mmHg) to receive either ALI 240 mg plus placebo matching the ALI/AML combination, or ALI/AML(240 mg/5 mg) along with a placebo matching ALI.
Blinding was maintained through standardized packaging. Each participant was provided with a large box containing a 5-week supply of medication, subdivided into five smaller boxes (each for 1 week). Each smaller box contained seven daily dose sachets. For ALI/AML group, each sachet contained one ALI/AML tablet (240 mg/5 mg) and one ALI-matching placebo tablet. For the ALI monotherapy group, each sachet contained one ALI tablet (240 mg) and one ALI/AML combination–matching placebo tablet. Identical packaging, color, appearance, smell, and labeling were employed across groups to ensure proper blinding.
For the ABPM sub-study, 40 participants were planned to be recruited. The eligibility criteria required participants to have uncontrolled blood pressure after 4 weeks of AML monotherapy, with a 24-hour mean ambulatory BP of ≥130/80 mmHg.
The study was approved by ethics committees of all participating centers and was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines. The trial was registered with ClinicalTrials.gov (NCT06465264).
Study Population
Patients were eligible for the study in the following conditions:
Primary inclusion criteria:
(1)
Patients aged 18 to 70 with mild-to-moderate essential hypertension;
(2)
Untreated patients (either newly diagnosed or with a history of hypertension who have not taken any antihypertensive medications for ≥ 2 weeks before screening) with msSBP 150 to < 180 mmHg and msDBP < 110 mmHg; patients with a history of irregular ALI treatment (240 mg once daily for < 4 weeks) or those who had missed doses for ≥ 5 days within the last 4 weeks, with msSBP 140 to <180 mmHg and msDBP < 110 mmHg; patients who had been on other antihypertensive therapies for ≥ 2 weeks with msSBP 140 to <180 mmHg and msDBP < 110 mmHg, deemed suitable for a switch to ALI 240 mg /day by the treating clinician; and patients who had been on ALI 240 mg/day for ≥ 4 weeks with msSBP 140 to <180 mmHg and msDBP < 110 mmHg;
(3)
Patients with msSBP 140 to < 180 mmHg and msDBP < 110 mmHg before randomisation;
(4)
Participants in the ABPM substudy with a 24-hour mean ambulatory blood pressure of ≥130/80 mmHg after 4 weeks of monotherapy.
Key exclusion criteria
(1)
Secondary hypertension;
(2)
MsSBP≥ 180 mmHg and/or msDBP≥ 110 mmHg, or a history of hypertensive emergencies or urgencies;
(3)
History of New York Heart Association (NYHA) Class III or IV heart failure, acute coronary syndrome, percutaneous coronary intervention, or other severe cardiac conditions such as cardiogenic shock, moderate-to-severe valvular heart disease, second-or third-degree atrioventricular block, bradycardia with a heart rate below 50 beats per minute, or severe arrhythmias within 6 months;
(4)
History of severe cerebrovascular events including hypertensive encephalopathy, cerebrovascular injury, stroke, or transient ischemic attack within the past 6 months;
(5)
Large aneurysm, aortic dissection, or dissecting aneurysm;
(6)
Renal artery stenosis or severe renal insufficiency;
(7)
Known allergies or intolerance to study drugs (such as angioedema);
(8)
Active viral hepatitis (including hepatitis B and hepatitis C), severe liver disease, or liver dysfunction;
(9)
Abnormal laboratory findings, including fasting blood glucose ≥ 11 mmol/L, creatinine > 1.5 times the upper limit of normal (ULN), potassium > 5.5 mmol/L, alanine aminotransferase (ALT) or aspartate aminotransferase (AST) > 2.5 times ULN, or total bilirubin (TBIL) > 2 times ULN. Additional details regarding eligibility criteria are provided in the Supplementary Material. All participants provided written informed consent before being included in the study.
Study EndpointEfficacy
Endpoints
Primary Endpoint
Change in msSBP at Week 12 post-randomisation.
Secondary Endpoints
(1)
Change in msDBP at Week12 post-randomisation;
(2)
Changes in msSBP and msDBP at Weeks 4 and 8 after randomisation;
(3)
Proportion in responders (msSBP/msDBP<140/90 mmHg, or a reduction in msSBP >20 mmHg and/or mean msDBP >10 mmHg) at Week12 after randomisation;
(4)
Target BP rates (msSBP/msDBP<140/90 mmHg) at Week 4, Week 8, Week 12, and Week 32 after randomisation as well as at the end of treatment.
Exploratory endpoints
Changes in 24-hour, morning, daytime, and nighttime mean ambulatory SBP/DBP at Week 12 after randomisation.
BP Measurements
Office BP measurements were conducted during the screening period, run-in period, and at Weeks 0, 4, 8, 12, 22, 32, 42, and 52 post- randomisation, following the hypertension management guidelines (Verification Regulation of Non-invasive Automatic Sphygmomanometer JJG 692-2010). The validated and calibrated upper-arm electronic blood pressure monitor (UA-651BLE-W, A&D Electronics Co., Ltd. Shenzhen, China) was utilized. In the morning, Office BP was measured twice at 1-2 minute intervals, with the mean value recorded; an additional measurement was performed if the difference between the two measurements of msSBP and/ or msDBP was ≥ 5 mmHg. Orthostatic BP should be measured after the last SBP measurement, with a one-minute delay after transitioning to an orthostatic position. Orthostatic hypotension was defined as a SBP reduction of ≥20 mmHg and/or a DBP reduction of ≥10 mmHg, irrespective of hypotension symptoms.
ABPM was also conducted during the screening, run-in periods, and at Week 12 post-randomisation using the AND TM-2430 device, following hypertension management protocols (Verification Regulation of Non-invasive Automated Sphygmomanometers JJG 692-2010). Before ABPM, clinic BP was measured in both arms; the higher BP arm was used, if the difference was ≥10 mmHg, otherwise, the non-dominant arm was selected. ABPM readings were taken every 20 minutes during the daytime awakening hours and every 30 minutes during nighttime sleeping hours. For ABPM validity, a minimum of 20 daytime readings, 7 nighttime readings, and 70% of the anticipated 24-hour readings (with at least one reading per hour) were required. The early morning period was defined as the initial 2 hours post-awakening, based on the participant’s schedule. The “awakening” and “sleeping” times were determined through participant self-reports. Changes from baseline in the morning, daytime, and nighttime SBP/DBP were analyzed. 40 participants were planned for the ABPM analysis.
The Shuoyun Integrated Information Management System (1.6.2.210422, Shanghai Shuo Yun Information Technology Co., Ltd. Shanghai, China) was used for data collection and reporting of both ABPM and office BP measurements.
Safety
Safety assessments included adverse events (AEs) and serious adverse events (SAEs), clinical laboratory tests (hematology, serum biochemistry, urinalysis), vital signs (excluding BP), physical examinations, 12-lead electrocardiograms(ECGs), and other relevant parameters. AEs and SAEs were documented from the time of consent. The laboratory tests were conducted during the screening period, the run-in period, as well as at Weeks 12, 32, and 52. Vital signs were measured during the screening period, run-in period, as well as at Weeks 0, 4, 8, 12, 22, 32, 42, and 52. Physical examinations were conducted during the screening period, run-in period, as well as at Weeks 12 and 52. 12-lead ECGs were performed during the screening period, run-in period, as well as at Weeks 4, 8, 12, 22, 32, 42, and 52.
Statistical Analysis
The sample size was calculated to detect a 5 mmHg difference in mean systolic blood pressure (msSBP) between ALI/AML and ALI, with 80% power, a standard deviation of 11 mmHg, and a one-sided significance level of 0.025 [22]. Accounting for a 20% dropout rate, 194 patients (97 per treatment arm) were required. Calculations were performed using PASS 16 software.
In this study, the intention-to-treat (ITT) analysis set includes all randomised subjects, regardless of whether they received the study medication. The full analysis set (FAS), based on the ITT principle, includes all subjects who were randomised, received at least one dose of the study drug during the double-blind period, and had at least one post-dose efficacy assessment. The PPS consists of subjects in the FAS who did not have any major protocol deviations and who provided data for the primary efficacy endpoint.
The primary efficacy endpoint analysis was analyzed using a mixed model for repeated measures (MMRM) on the FAS with supplementary analyses on the PPS. The model incorporated changes in msSBP at each visit during the double-blind treatment period as the dependent variable. The model included treatment group, time, and group-time interactions as fixed effects, and baseline msSBP as a covariate. The difference in least squares mean (LSM), 95% confidence intervals (CIs), and P values were calculated. Additionally, subgroup analyses based on randomisation stratification and other baseline characteristics were conducted.
The secondary endpoints of changes in msSBP and msDBP were evaluated with MMRM on FAS and PPS, and target BP rates or response rates at Week 12 post-randomisation were analysed using a logistic regression model. The target BP rates at Week 32 and at the end of treatment were analysed using descriptive statistics.
The baseline characteristics were summarized using descriptive statistics on the FAS, and the safety assessments were conducted on the safety set (SS). All statistical analyses were conducted using SAS version 9.4 software (SAS Institute, Inc.).



Comments (0)