Cell and viruses
HEK293T cells (CRL-3216, ATCC) and HEp-2 cells (CCL-23, ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1× penicillin-streptomycin (Thermo, USA) at 37 °C, 5% CO2. RSV Long (VR-26) and RSV B1 were passaged in HEp-2 cells in DMEM/2% FBS, purified via ultrafiltration and stored in DMEM/3% sucrose at −80 °C as the previous reported47.
Design of PreF-EABR constructs
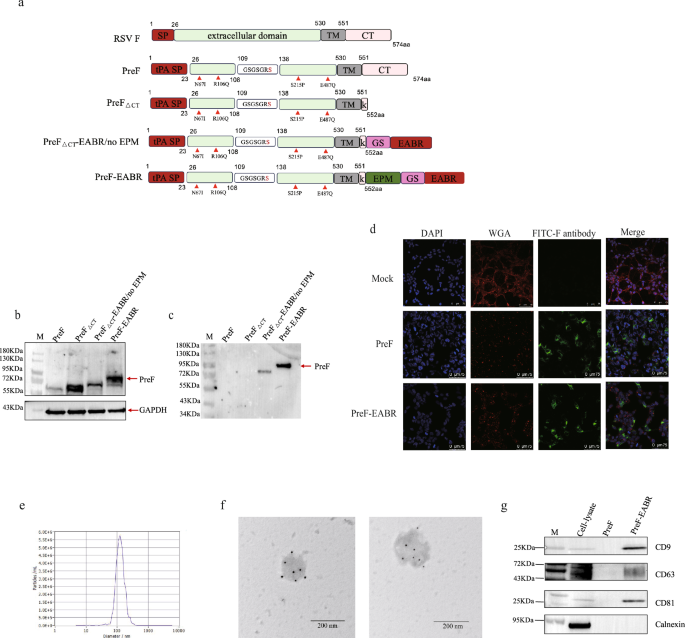
The amino acid sequence of the RSV fusion protein used was RSV A CC14-10 (GenBank: APW77972). PreF construct was codon-optimized and modified by introducing amino acid substitutions (N67I, R106Q, F137S, S215P, and E487Q), the P27 (109-136aa) was substituted with the linker (GSGSGR)47,48, and the native signal peptide was replaced with tPA signal peptide. The C terminal 23 residues was truncated to optimized cell surface expression to form PreF△CT construct22.PreF△CT-EABR/no EPM construct was generated by inserting the EABR domain (160-217aa) of the human CEP55 protein in C terminal to the PreF△CT construct separated by the GGGGS linker. An endocytosis prevention motif (EPM), a 47-residue insertion from the murine Fc gamma receptor FcgRII-B1 cytoplasmic tail was used to prevent coated pit localization and endocytosis, inserting the upstream of the GS linker and the EABR domain and behind the PreF△CT to form PreF-EABR construct. All constructs were cloned into the pCDNA3.1 expression plasmid.
Production of PreF-EABR
PreF-EABR was generated by transfecting HEK293F cells through the X-treme GENETM HP transfection reagent (Roche, Switzerland), 48 h post-transfection, cells were centrifuged at 800 × g for 10 min to separate the cell pellet and supernatant. The cell supernatant was processed according to the previously reported method to obtain eVLPs21. In brief, supernatant was filtered through 0.45 μm filter to further remove the cell debris, and then, eVLPs was precipitated by 20% sucrose density gradient centrifugation at 50000 rpm for 2 h at 4 °C. The pellet was resuspended in PBS at 4 °C overnight, and then samples were centrifuged at 10000 × g for 10 min to remove residual cell debris.
Western blot (WB)
Cell pellet was lysed by the RIPA lysis buffer and eVLPs were prepared for SDS-PAGE, then transferred onto PVDF membranes for WB analysis. After blocking with 5% skim milk. For F-specific WB, the membranes were incubated with mouse anti-F monoclonal antibody (1:1000 dilution, Abcam, USA) overnight at 4 °C followed by HRP-conjugated goat anti-mouse IgG antibody (1:5000 dilution, Abcam, USA). For identification of extracellular vesicle, membranes were separately incubated with anti-CD63 antibody (1:1000 dilution, Abcam, USA), anti-CD9 antibody (1:1000 dilution, Abcam, USA), anti-CD81 antibody (1:1000 dilution, Abcam, USA), anti-Calnexin antibody (1:1000 dilution, Abcam, USA) overnight at 4 °C followed by HRP-conjugated goat anti-mouse IgG antibody (1:5000 dilution, Abcam, USA).
Immunofluorescence Assay (IFA)
The slides of cells were sterilized by 75% ethanol and PBS rinsing and air-dried in 6-well plates. Then, HEK293 cells were seeded at 2 × 10⁵ cells/well and cultured in DMEM with 10% FBS at 37 °C, 5% CO2. When the cell density reached 50-70% confluence, transfection complexes were prepared, which consist of 2.5 μg plasmid DNA (PreF or PreF-EABR construct) and 5 μl X-treme GENETM HP transfection (Roche, Switzerland) in 180 μl Opti-MEM (Gibco, USA) were then added to the cells. 24 h post-transfection, cells were subjected to a series of processing steps: fixed with 4% paraformaldehyde (Sigma, Germany) for 20 min. After PBS washed, cells were stained with 5 μg/ml WGA-633 (Thermo Fisher, USA) for 5 min, then cells were permeabilization using 0.2% Triton X-100 (Sigma), and nuclear counterstaining with 1 μg/ml DAPI (Sigma, Germany). Subsequently, the cells were stained with FITC-anti F antibody (1:400, Vazyme, China) for 30 min. After PBS washed, the images of the targeting proteins were viewed and analyzed using confocal microscopy (LEICA TCS SP8, Germany).
Characterization of PreF-EABR
The number and size of eVLPs were measured by nanoparticle tracking analysis (NTA) using a ZetaView_Particle Metrix (PMX-120, Germany). Immunotransmission electron microscopy (IEM) was used to identify the particle. eVLPs were fixed with an equal volume of 4% paraformaldehyde, and then pipette the liquid onto a carbon-coated nickel grid, block with 0.05 M glycine and then block with 1% BSA (Sigma, Germany), incubate with rabbit anti F antibody overnight at 4 °C. After washed, incubated with 6-nm colloidal gold Goat anti rabbit antibody for 3 h. Fixed with 2.5% glutaraldehyde, then stained with 2% uranyl acetate. IEM was performed using a JEOL–1200 electron microscope (JEOL, Japan) operated at 80 KV to analysis.
mRNA vaccine preparation
PreF and PreF-EABR gene were codon optimized through LinearDesign and insert in-vitro transcription vector, which flank with the 5’ and 3’ untranslated regions and a 120 nt poly(A) tail. After the plasmid was linearized by digestion with Bsa I, T7 RNA polymerase-mediated transcription was employed to synthesize the mRNA using the T7 High Yield RNA Transcription Kit 2.0 (N1-Me-pUTP) (Novoprotein, China), After this, transcription products were incubated at 37 °C for 1 h by treatment with DNase I. RNA was capped using Cap 1 Capping System Kit (Novoprotein, China) and purified using oligo dT (BIA, Germany). Quality of mRNA was analyzed by agarose gel electrophoresis and stored at -80 °C. For mRNA encapsulation into LNP, the LNP were prepared using Moderna’ SM102 LNP formulation and encapsulated by Novoprotein (Shanghai, China). Vaccine formulation was characterized for particle diameter, polymer dispersity index (PDI), and zeta potentials using NanoBrook Omni ZetaPlus (Brookhaven, Germany).
Immunization and virus challenge
Six- to eight-week-old female Balb/c mice were purchased from Beijing Vital River Laboratory Animal Technology Co. (Beijing, China) and housed in appropriate animal facilities at the laboratory animal center of the Chinese Center for Disease Control and Prevention. All animal experiments were conducted under the approval of the ethics committee of the Chinese Center for Disease Control and Prevention (approval NO.20221107115).
Mice (n = 5/group) were immunized intramuscularly (i.m.) on days 0 and 14 with PreF mRNA or PreF-EABR mRNA at doses of 0.5 μg, 1 μg, or 2.5 μg in a 100 μl volume (50 μl injected into each thigh muscle). LNP was used as control. Serum samples were collected for Ab response and RSV-specific neutralization assay analysis at different time points. For the challenge experiments, Balb/C mice (n = 5/group) were vaccinated i.m. with LNP, PreF mRNA, and PreF-EABR mRNA at the dose of 2.5 μg in a 100 μl volume (50 μl injected into each thigh muscle) with an interval of 2 weeks. FI-RSV were used as positive control at the dose of 20 μg. At the same time, mice that were neither immunized nor challenged were used as the negative control group (naïve group). Four weeks after the last vaccination, mice were anesthetized with isoflurane and then intranasally challenged with 2 × 106 PFU of RSV Long in a 30 μl volume. Five days post - challenge, the mice were euthanized by cervical dislocation, and their lung tissues and nasal turbinates were collected and stored at −80 °C until processed.
For GC B and Tfh immune response, Balb/C mice (n = 5/group) were immunized with LNP, PreF mRNA or PreF-EABR mRNA at the dose of 2.5 μg in a 100 μl volume (50 μl injected into each thigh muscle). At the end point of experiments, mice were euthanized by cervical dislocation and spleens and draining lymph nodes (dLN) were collected and processed to obtain single-cell suspension for analysis at 7 and 14 days post-last vaccination.
For the RNA-Seq experiments, BALB/c mice (n = 8/group) were vaccinated i.m. with LNP, PreF mRNA, and PreF-EABR mRNA at the dose of 2.5 μg in a 100 μl volume (50 μl injected into each thigh muscle). Naïve mice were set as the control. PBMCs were isolated 24 h post-injection for bulk RNA-seq (n = 3) and mouse serum were collected for cytokine array (n = 5).
Measurement of PreF-specific IgG
PreF protein (SinoBiological, China) were coated into 96-well plates (Corning, China) at a concentration of 50 ng/ well and incubated overnight at 4 °C. The plates were washed with PBS containing 0.01% Tween-20 (PBST) and blocked for 2 h at 37 °C with 5% skim milk. Sera samples serially diluted were added and incubated for 1 h at 37 °C. binding IgG were determined using HRP-conjugated goat-anti-mouse IgG (1:30,000, Abcam, USA) for 1 h at 37 °C, respectively. TMB substrate (Solarbio, China) was used for development and the absorbance was read at 450 nm and 630 nm wavelength. The endpoint titer was calculated as previous reported47.
RSV-specific neutralization assay
A micro-neutralization assay was used to determine the anti-RSV Long and RSV B1neutralizing antibody titers, as our previously described47. The neutralization titers 50 (NT50) were defined as the equivalent of the serum dilution required for 50% neutralization of viral infection.
Histopathological analysis
After the RSV challenge, lungs were immersed in 4% paraformaldehyde and stained with hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) for pathological evaluation and mucus secretion. Four parameters associated with lung pathological changes were evaluated. Each of these parameters was scored separately as previously described49.
RSV titer in lungs and nasal turbinates
The RSV Long loads in the lung and nasal turbinates were quantified using qRT-PCR as previous reported47. After homogenization of lung tissue and nasal turbinates, the virus RAN was isolated and reverse transcription PCR were performed using HiScript II One Step qRT-PCR Probe Kit (Vazyme, China). Viral loads were calculated using a standard curve. The primer and probe sequences were as follows.
L qPCR Forward: 5′-GAACTCAGTGTAGGTAGAATGTTTGCA-3′.
L qPCR Reverse: 5′- TTCAGCTATCATTTTCTCTGCCAAT- 3′.
L-probe: FAM-ATTTGAACCTGTCTGAACATTCCCGGTTGCAT- BHQ1.
PBMCs, Splenocyte, dLN and LLPC cell preparation
PBMCs were isolated using LymphoprepTM in SepMate™ PBMC Isolation Tubes according to the manufacturer’s instructions. Blood collection involved 400−500 μl of peripheral blood sampled via the buccal vein. Spleens were homogenized and passed through 40 μm cell strainers to produce single cells. LNs (Inguinal LNs) were harvested after immunization, then homogenized and passed through 40 μm cell strainers to produce single cells. LLPCs were isolate from bone marrow (BM) of mouse femur using a syringe.
Folw cytometry detection
Erythrocytes were lysed in RBC lysis buffer (eBioscience, USA). Cells were washed with DPBS, then blocked with anti-CD16/CD32 antibody (BD, USA), and stained with Live/Dead staining Kit (Invitrogen, USA) in DPBS.
Effector T cell response
PBMCs (1 × 106 cells/100 μl) were plated in 96-well V tissue culture plates and stimulated with RSV F peptide pools (A total of 130 peptides spanning the full-length F protein of RSV were synthesized as 15-mers, with 11 overlapping amino acids) and soluble co-stimulatory molecules CD28 and CD49d at a final concentration of 2 μg/mL and 1 μg/mL, and cultured at 37 °C in 5% CO2 for 5 h, followed by the addition of Golgi-Plugs (BD Biosciences) at 37 °C with 5% CO2 for another 6 h. After blocking and staining with the LIVE/DEAD, cells were stained with Percp/Cy5.5-CD3 (1:400, BD, USA), FITC-CD4 (1:400, BD, USA), and PE/Cy7-CD8a (1:400, BD, USA) at 4 °C for 0.5 h, then permeabilized with fixation/permeabilization solution (Invitrogen, USA). Samples were then intracellularly stained by incubating with BV421- TNF-α (1:250, BD, USA), PE-IL-2 (1:250, BD USA), APC-IFN-γ (1:250, BD, USA) or BV421-IL-4 (1:250, BD, USA), PE-IL-13 (1:250, BD, USA), and APC-IL-5 (1:250, BD, USA) for 1 h at 4 °C. Cells were washed and resuspended in 200 μL of PBS buffer for acquisition using BD Arial II (BD Bioscience), and analyzed using FlowJo v10. The gating strategy is shown in Fig. S1.
GC B and Tfh staining
LNs and splenocytes were stained with BV421-CD4 (1:250, BD, USA), PE/Cy7-Fas (1:250, BD, USA), APC-GL7 (1:250, BD, USA), BV786-B220 (1:250, BD, USA), FITC-RSV F protein (1:100, Vazyme, China), PerCP/Cy5.5-CXCR5 (1:250, BD, USA), PE-PD-1 (1:250, BD, USA), PE/Dazzle594-CD19 (1:250, BD, USA), and APC-Cy7-IgD (1:250, BD, USA) for 1 h. The gating strategy is shown in Fig. S2.
MBC and LLPC staining
Splenocytes and LLPC were stained with BV421-CD138 (1:250, BD, USA), PE-B220 (1:250, BD, USA), FITC-RSV F protein (1:100, Vazyme, China), PerCP/Cy5.5-CD19 (1:250, BD, USA), PE/Cy7-CD38 (1:250, BD, USA), APC-GL7 (1:250, BD, USA) and APC/Cy7-IgD (1:250, BD, USA) for 1 h. The gating strategy is shown in Fig. S3
RNA-seq analysis
RNA-seq and differential expression analysis were performed at Novogene (Beijing, China). After quality control, differentially expressed genes (DEGs) were defined as those exhibiting ≥2-fold up- or downregulation (|log₂ (fold change) | > 1, q < 0.05) in vaccinated versus naïve mice or versus vaccinated PreF mRNA group. Pathway analysis of significantly differentially expressed genes identified from the RNA-seq results was performed using KEGG in R Studio v.3.0.3. Pathways and biological processes with adjusted P-value ≤ 0.05 were considered significant.
Multi-cytokine assay
Mice serum was collected at 24 h post prime immunization for a multi-cytokine assay. Cytokine levels were evaluated with a Luminex 200 system using a panel of 23 mouse cytokines (LX-MultiDTM-23) according to the manufacturer’s instruction. The selected cytokines included Eotaxin/CCL11, G-CSF, GM-CSF, IFN-γ, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-17A, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, GRO-α(Gro-a/KC/CXCL1), MCP-1/CCL2, MIP-1α/CCL3, MIP-1β, RANTES, TNF-α.

Comments (0)