
Remember me


Across the studied concentration range (1–300 µM), icotrokinra exhibited low permeability that was not saturable in polarized cell monolayer models expressing MDR1, the gene encoding human P-glycoprotein (P-gp), with apparent permeability coefficient (Papp) (A-B) ranging from 0.90 to 1.96 nm/s and Papp (B-A) from 0.61 to 1.98 nm/sec (Supplementary Table S1). Efflux ratios (ERs) were < 2 across the concentration range in the absence (0.7–1.2) or presence (1.1–1.3) of the P-gp inhibitor, valspodar, confirming no directional differences in permeability and no change in permeability with P-gp inhibition, indicating that icotrokinra is not a substrate of P-gp. Under the same incubation conditions, ERs of [14C]-icotrokinra were also < 2 in mock-transfected cell monolayers.
Plasma Protein Binding and Blood Cell PartitioningIcotrokinra showed low protein binding across species (Supplementary Table S2), with mean percent bound ranging between 43.7% and 56.0% in mouse, rat, and monkey plasma and 49.7–55.2% in human plasma. Protein binding was notably higher in rabbit plasma (mean bound compound 70.3–71.9%). No concentration-dependent differences in protein binding were observed across the tested concentrations (0.01–1 µM). Additionally, icotrokinra was found to be stable in plasma at 37°C for up to 16 h. Mean percent binding values ranged from 25.7% to 35.9% for 4% human albumin, 1.07% to 10.6% for 0.07% AAG, and 7.56% to 8.53% for 0.14% AAG, whereas in mixtures of 4% albumin with 0.07% or 0.14% AAG, percent binding ranged from 25.8% to 37.1% and 20.0% to 33.8%, respectively. Thus, icotrokinra appeared to be preferentially binding to human serum albumin as compared with AAG. Since plasma protein binding is low, no DDI related to plasma protein binding are expected.
Icotrokinra exhibited low blood partitioning and no preferential binding to RBCs, with mean observed KWB/P and KRBC/P values of < 1 in rat, dog, monkey, and human whole blood (Supplementary Table S3). Across icotrokinra concentrations of 2.5–250 ng/mL, mean KWB/P values ranged from 0.63 to 0.84 in rat, 0.61 to 0.84 in dog, 0.43 to 0.79 in monkey, and 0.49 to 0.61 in human whole blood after 2 h of incubation.
Metabolic StabilityIcotrokinra demonstrated in vitro stability in feces, GI mucosa, and hepatocytes across rat, monkey, and human species. At 50 µM, the mean half-life (t1/2) values in fecal and GI mucosal matrices exceeded 24 h for all three species. After 24 h, the percentages of icotrokinra remaining in fecal homogenates relative to the initial concentrations were 81.3%, 75.2%, and 76.0% in rats, monkeys, and humans, respectively (Supplementary Figure S1a). The respective percentages remaining in GI tissues at 24 h ranged between 92.2% and 100% across species (Supplementary Figure S1b). Icotrokinra was also shown to be stable in SGF alone and solutions containing purified GI enzymes, including chymotrypsin, pancreatin, pepsin, and trypsin, with mean t1/2 values exceeding 24 h and percentages of icotrokinra remaining after 24 h ranging between 84.4% and 100% across matrices (Supplementary Figure S1c). Icotrokinra turnover was low when incubated with hepatocytes, with mean t1/2 > 371 minutes (min) (> 6 h) and intrinsic clearance (CLint) < 1.8 µL/min/106 cells. After 2 h of hepatocyte incubation, icotrokinra remained largely unchanged, with 95.7%, 92.8%, and 96.6% of the drug still present, in rat, monkey, and human hepatocytes, respectively, suggesting that it is not subject to significant hepatic metabolic clearance (Supplementary Figure S1d).
Drug–Drug Interaction StudiesIcotrokinra was evaluated for its potential as a substrate and inhibitor of human transporters. Icotrokinra was not a substrate for P-gp, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, MATE2-K, and BCRP and did not inhibit P-gp, OAT1, OAT3, OCT2, MATE1, MATE2-K, BCRP, or BSEP activity (Supplementary Table S1; Supplementary Table S4; Supplementary Table S5; Supplementary Table S6). At the highest tested concentrations (measured concentrations of 24.7 µM for OATP1B1 and 24.2 µM for OATP1B3), 43.9% and 48.0% inhibition of OATP1B1 and OATP1B3 activity, respectively, were observed (Supplementary Table S4). No interaction with these transporters is anticipated at clinically relevant concentrations (the maximum plasma concentration [Cmax] for a 200 mg dose is approximately 0.002 µM), suggesting icotrokinra would not influence the PK of drugs that are substrates of these transporters.
Icotrokinra (up to 100 µM) did not exhibit concentration-dependent inhibition of major human CYP enzymes. The half-maximal inhibitory concentration (IC50) for all CYP isoforms was ≥ 100 µM, indicating minimal potential for CYP-mediated DDIs. At the highest concentration tested (100 µM), maximum inhibition was < 10% for most CYP isoforms, with only CYP1A2 (19%) and CYP2E1 (30%) showing weak inhibition. These findings suggest icotrokinra is not a significant inhibitor of the selected CYP enzymes under the conditions tested.
The potential for icotrokinra to induce CYP1A2, CYP2B6, and CYP3A4 mRNA expression was evaluated in cultured human hepatocytes at concentrations of up to 90 µM. Icotrokinra caused no induction (< 2-fold change in mRNA expression) and minimal response relative to the positive control (< 5% at 90 µM; Supplementary Table S7), indicating no significant induction of the selected CYP enzymes under the conditions tested. Together with the hepatocyte stability and CYP inhibition results, these findings indicate icotrokinra has a low potential for enzyme-mediated DDIs.
In Vivo Pharmacokinetic Studies of IcotrokinraPharmacokinetic Profile in Rats and MonkeysFollowing a single IV dose (2 mg/kg in rats, 1 mg/kg in monkeys), icotrokinra exhibited moderate distribution, with estimated volumes of distribution at steady state (Vss) of 459 mL/kg in rats and 299 mL/kg in monkeys. The area under the plasma concentration–time curve from time 0 extrapolated to infinity (AUC0–∞) was 3810 ng*h/mL in rats and 12,000 ng h/mL in monkeys (Table 1). Plasma elimination t½ was 0.762 h and 3.47 h in rats and monkeys, respectively, and mean clearance was 8.77 mL/min/kg and 1.44 mL/min/kg, respectively.
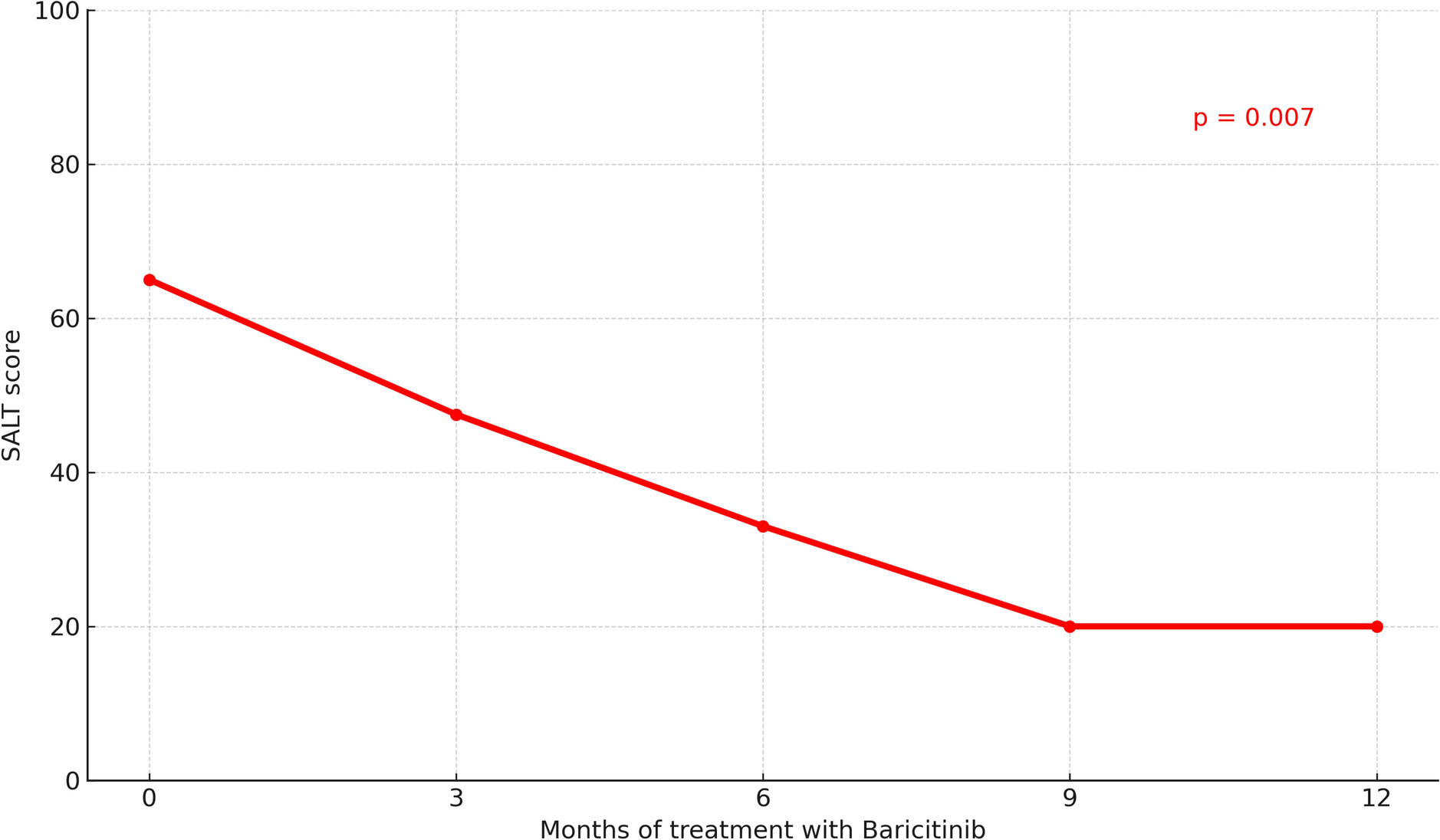
Table 1 Plasma pharmacokinetics of icotrokinra in rats and monkeys following intravenous or oral dosingOral administration of icotrokinra at 20, 30, 100, and 300 mg/kg in rats yielded a mean Cmax of 8.68, 134, 76.3, and 134 ng/mL; oral bioavailability (F) of 0.12%, 0.32%, 0.17%, and 0.11%; and an AUC0–∞ of 44.2, 181, 317, and 1,120 ng*h/mL (Table 1). Terminal half-life was approximately 2 h where estimatable. Similar results were observed in monkeys following single oral doses of 2.5, 7.5, or 22.5 mg/kg, with plasma concentrations peaking 1–2 h post-dose (Table 1, Fig. 2); respective mean Cmax values were 11.0, 27.8, and 84.3 ng/mL, demonstrating an approximately dose-proportional increase. Systemic exposure (AUC0–∞) increased dose-proportionally by 2.75-fold (7.5 versus 2.5 mg/kg) and 9.88-fold (22.5 versus 2.5 mg/kg) across tested dose levels. Oral bioavailability of icotrokinra in monkeys was low, with values of 0.27%, 0.25%, and 0.30% at doses of 2.5, 7.5, and 22.5 mg/kg, respectively. However, even at the lowest dose of 2.5 mg/kg, icotrokinra plasma concentrations exceeded its IC50 for inhibition of IL-23-induced signal transducer and activator of transcription 3 (STAT3) phosphorylation in human peripheral blood mononuclear cells (PBMCs; Fig. 2) [12]. Elimination t½ was consistent across the oral dose groups, ranging from 5.6 h to 6.4 h (Table 1).
Fig. 2
Plasma concentrations of icotrokinra measured over 24 h in monkeys following a single oral administration. Dotted line represents the icotrokinra IC50 for IL-23-induced STAT3 phosphorylation in human PBMCs after 24 h [12]. SD values below 0 are not plotted. h hour, IC50 half-maximal inhibitory concentration, IL interleukin, PBMC peripheral blood mononuclear cell, SD standard deviation, STAT3 signal transducer and activator of transcription 3
Tissue Distribution in MonkeysIn monkeys, IV-administered icotrokinra (1 mg/kg) was found to be well distributed to tissues relevant to psoriasis, psoriatic arthritis, and IBD, including skin, joints, and GI tissues (Fig. 3). Skin, blister fluid, ileum, cecum, colon, rectum, patellar synovium, patellar tendon, intervertebral ligament, Achilles tendon, and sacroiliac ligament had estimated tissue concentrations relative to plasma (tissue:plasma) between 45.8% and 156%. Risankizumab, a monoclonal antibody that inhibits the IL-23 cytokine, showed tissue concentrations relative to serum (tissue:serum) ranging from 4.07% to 32.1% after a 1 mg/kg IV dose in monkeys, using the same testing site and collection procedures, with timepoints adjusted accordingly for the peptide and antibody (Fig. 3).
Fig. 3
Tissue distribution of icotrokinra (1 mL/kg intravenous) and risankizumab (2 mL/kg intravenous) in monkeys. Icotrokinra tissue concentration presented as tissue:plasma percentage. Risankizumab tissue concentration presented as tissue:serum percentage
Metabolic Profiling in Rats and MonkeysFollowing oral administration of [14C]-icotrokinra at 300 mg/kg in rats, unchanged icotrokinra was the predominant species (> 95%) in all plasma samples. Eight metabolites (M6, M7, M8, M9, M12, M25, M26, and M27), along with unchanged icotrokinra, were identified in urine at comparable levels (Table 2). In feces, unchanged icotrokinra was the dominant species (96.3%), with minor contributions from metabolites M14 (2.2%) and M16 (1.5%). The recovery of unchanged drug from feces (0–24 h) was on average 72.8% of the total dose (Table 2). M27 and M16 were detected in the spiked blank plasma, feces, and urine samples, indicating that the presence of these metabolites could be partially attributed to sample processing.
Table 2 Mass balance of [14C]-icotrokinra (300 mg/kg) and its main metabolites in rats (~ 400 µCi/kg) and monkeys (~ 100 µCi/kg) after oral administrationFollowing a single oral dose of [14C]-icotrokinra at 300 mg/kg in monkeys, unchanged drug was the predominant species in plasma, and the main species recovered in the excreta, corresponding to 64.2% of dose recovery. In feces, metabolites (M5, M6, M8, M10, M12, M13, and M14) accounted for very low amounts of the oral dose (Table 2). Four metabolites (M7, M9, M11, and M14) and unchanged icotrokinra were detected in urine at similarly low levels (Table 2).
Excretion in Rats and MonkeysFollowing an oral dose of 300 mg/kg [14C]-icotrokinra, fecal excretion was the primary route of elimination in both rats (96.8% recovered) and monkeys (67.0% recovered) within 96 h, with the majority eliminated in the first 24 h (Supplementary Table S8). Total recovery of the dose over 96 h, including from urine, feces, and cage washing, was 97.3% in rats and 73.1% in monkeys. In both rats and monkeys, < 1% of icotrokinra was recovered from urine within 96 h; however, this likely represents a major clearance pathway for systemically absorbed drug since the oral bioavailability is similarly low. Quantitative whole-body autoradiography of a single monkey carcass revealed that icotrokinra levels were below the lower limit of quantification (LLOQ) in all tissues at 96 h post-dose, indicating negligible retention.
Icotrokinra Phase 1 Clinical Studies in Healthy VolunteersParticipant Disposition and DemographicsBaseline characteristics of healthy adults enrolled in the FIH study are presented in Table 3. All participants were men. In the SAD population, the median participant age was 23.0 years; 51.3% were white; and median weight and BMI were 73.5 kg and 23.7 kg/m2, respectively. The median participant age in the MAD population was 28.0 years; 66.1% were white; and the median weight and BMI were 79.5 kg and 25.2 kg/m2, respectively. In the MAD population, two participants withdrew consent for reasons unrelated to safety findings: one participant withdrew consent for participation and the other discontinued due to noncompliance.
Table 3 Baseline characteristics in Phase 1 healthy volunteersAmong the 24 healthy participants enrolled in the relative bioavailability study, 23 completed the study as planned; 1 participant discontinued the study due to a treatment-emergent AE (otitis media). The median age was 55.0 years, 79.2% were men, 20.8% women, and 95.8% were white (Table 3). The median weight and BMI were 79.7 kg and 26.0 kg/m2, respectively.
Pharmacokinetic Profile in HumansLimited PK of icotrokinra in humans has been previously reported for the SAD and MAD populations [12]. In the SAD population, following single oral dose administration of icotrokinra formulated as a solution, rapid absorption was observed, with a median time to reach maximum plasma concentration (tmax) ranging from 1.5 h to 4.0 h post-dose (Table 4). The mean t1/2 ranged from approximately 9–12 h across dose cohorts (25–1000 mg). The median plasma concentrations of icotrokinra increased with increasing doses up to 1000 mg (Fig. 4A). In the MAD cohort, following multiple doses of icotrokinra formulated as a solution, the median tmax ranged from 1.5 h to 5.0 h on Day 1 and from 1.0 h to 6.0 h on Day 10 (Table 4). The mean t1/2 was approximately 9–16 h. Consistent with the half-life, a comparison of Cmax and AUC from time 0 to the time of the last measurable concentration (AUC0–last) values on Days 1 and 10 indicated an accumulation ratio of 0.70–1.65-fold for Cmax and 0.9–1.49-fold for AUC with once-daily multiple dosing. Also following multiple doses, the median plasma concentrations on Days 1 and 10 exhibited a dose-dependent increase (Fig. 4B). A dose-proportional increase was also seen for both mean Cmax (Fig. 5A) and AUC from time 0 h to 24 h (AUC0–24 h; Fig. 5B). The slopes (linear coefficients) for Cmax and AUC0–24 h on Day 1 and Day 10 were close to unity, and the 90% CIs included unity.
Table 4 Summary of icotrokinra PK parameters in Phase 1 healthy volunteersFig. 4
Plasma concentrations of icotrokinra in healthy participants following a single dose (A) or multiple doses at Days 1 and 10 (B) of the solution formulation, and comparing 1 × 200 mg (Phase 3) tablet with 2 × 100 mg (Phase 2) tablets (C). *Data below the level of quantification are not plotted. The 1000 mg dose was administered for 8 days only. h hour
Fig. 5
Maximum plasma concentration (A) and area under the plasma concentration–time curve at 24 h (B) following multiple doses of icotrokinra in healthy participants. Dose proportionality was assessed using the power model with natural log-transformed PK parameter values and natural log-transformed dose (excluding the placebo group). Line represents best fit on the basis of linear regression analysis. AUC024h area under the curve at 24 h, Cmax maximum plasma concentration, ln natural log, PK pharmacokinetic
In the relative bioavailability study, a median tmax of 2 h was observed following a single oral dose of 200 mg icotrokinra, formulated as 1 × 200 mg (Phase 3) or 2 × 100 mg (Phase 2) tablets, under fasted conditions (Table 4). The mean t1/2 was also consistent across formulations, with values ranging from 12.4 h to 13.0 h. After reaching a maximum, plasma concentrations of icotrokinra exhibited a biphasic decline with both the 1 × 200 mg (Phase 3) and 2 × 100 mg (Phase 2) tablets (Fig. 4C). Point estimates of the geometric mean ratios (GMRs) indicated that the relative bioavailability of icotrokinra for the 1 × 200 mg (Phase 3) tablet compared with the 2 × 100 mg (Phase 2) tablets was 88.1% for Cmax, 94.2% for AUC0–last, and 94.9% for AUC0–∞ (Table 5).
Table 5 Comparison of icotrokinra PK parameters following single administration of 200 mg icotrokinra in Phase 1 healthy volunteers in the relative bioavailability studyMetabolic Profiling and Excretion in HumansUnchanged icotrokinra was the only drug-related material detected in plasma across both the SAD and MAD populations. Urinary excretion of unchanged icotrokinra was negligible (< 0.001%), while fecal excretion of unchanged icotrokinra increased with dose, ranging from 37.2% at 10 mg to 80.7% at 1000 mg. No metabolites were detected in plasma or urine. Detectable concentrations of icotrokinra were noted in sigmoid colon and rectal biopsy samples collected from participants following 25 mg and 100 mg icotrokinra doses.
Overall, no SAEs were reported and findings from safety assessments in the FIH were as those reported previously [12].
Comments (0)