Chemotherapeutic drug treatment in vitro assay
The wild-type (WT) cell lines of U2OS, 143B, U87-MG and A549 were seeded in 12-well plates with or without round coverslips. The next day, cisplatin, doxorubicin, etoposide, and methotrexate in appropriate concentration were added in culture as needed or proper vehicle solution for 6 h. The cells were treated appropriately for WB, fluorescence-activated cell sorting, and IF.
Immunofluorescence staining (IF)
In this study, the indicated cells were plated on round coverslips in 6-well plates, treated with drugs, and fixed with 4% poly formaldehyde at room temperature (RT). The cell coverslips were washed three times with 1x TBST buffer, with each washing 5 min. The coverslips were incubated at RT with approximately 100 μL of blocking and permeabilization buffer for 1 h. The blocking and permeabilization buffer were removed, and the primary antibodies anti-G3BP1 and anti-TIAR (both are markers of SGs) diluted in an antibody dilution buffer (~100 μL, dilution: 1:50) were added to the coverslips and incubated overnight at 4 °C. On the following day, the primary antibody solution was removed, and the tissue slides were quickly washed three times using 1x TBST buffer in the staining dish, with each wash lasting 5 min. The coverslips were incubated at RT with approximately 100 μL of secondary antibodies (1:500 dilution) and DAPI (1:10 000 dilution), both diluted in an antibody dilution buffer, for 1.5 h. The secondary antibody/DAPI solution was removed, and the coverslips were quickly washed three times using 1x TBST buffer in the staining dish, with each wash lasting 5 min. For mounting, the slides were applied approximately 50–100 μL of mounting medium to each slide and covered with a square coverslip. The slides were allowed to dry overnight in a dark environment. The next day, the coverslip was sealed to the edge of the slide using transparent nail polish, which was left to dry for 1 h. Finally, the slides were imaged under a confocal microscope.
SG quantitative analysis
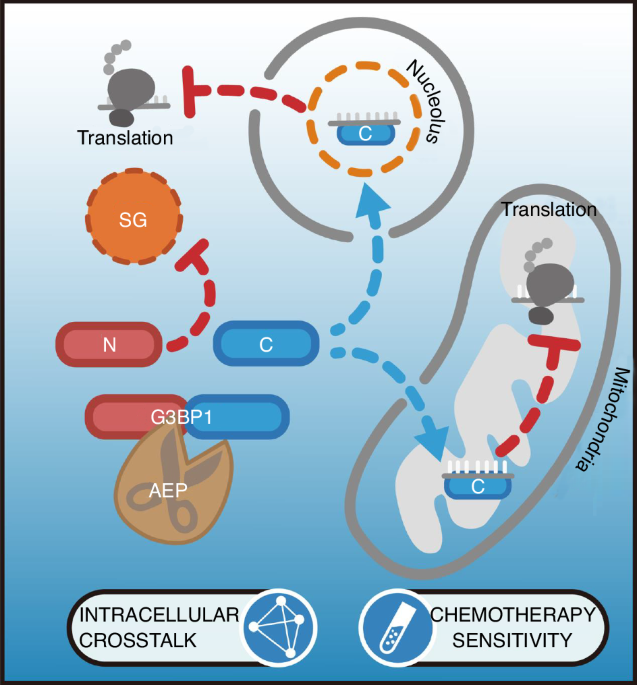
The chemotherapy drug-induced SG were visualized via IF with anti-G3BP1 and anti-TIAR antibodies. SGs were quantified with the SG counts per cell and the SG-positive cell count ratio.
WB
The lysates of the indicated cells or tissue were prepared in RIPA buffer (Cat# R0020, Solarbio, Beijing, China) containing phenylmethylsulphonyl fluoride (Cat# 329-98-6, Sigma-Aldrich) and Complete Protease Inhibitor Cocktail (Cat# 4693116001, Roche, Basel, Switzerland). Lysates were incubated for 30 min on ice and centrifuged for 10 min at 12 500 × g and 4 °C. A BCA protein assay kit (Cat# 23225, Thermo Fisher Scientific) was used to determine the protein concentrations. Sample proteins were separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis and transferred to PVDF membranes. After blocking with TBST (TBS with 0.1% Tween 20) containing 5% skim milk, the membranes were incubated with primary antibodies overnight at 4 °C. The membranes were then incubated with appropriate HRP-conjugated secondary antibodies at RT for 1 h. A ChemiDoc Imaging System (Cat# 12003154, ChemiDoc™ MP Imaging System, Bio-Rad, Hercules, CA, USA) was used to detect chemiluminescence.
Co-immunoprecipitation (IP) and mass spectrometry (MS) analysis
AEP-KO U2OS infected with human AEP-expressing lentivirus were treated with 50 μmol/L cisplatin or vehicle for 6 h. Cells were lysed in IP buffer (Cat# 87787, Thermo Fisher Scientific) and then subjected to IP using an anti-AEP antibody (1:50 dilution). A portion of the immunoprecipitate was used for silver staining, and the remaining immunoprecipitate was sent to Oebiotech Co., Ltd. (Shanghai, China), for LC-MS/MS analysis.
MS analysis of AEP-KD/tG3BP1-C rescue U87-MG cells were performed in a similar process. Cell lysates were subjected to IP using an anti-Flag antibody (1:100 dilution).
AEP cleaved G3BP1 in vitro
Recombinant human G3BP1 (10 μg) (WT, N258A, and N309A point mutants) was incubated with recombinant human AEP or in 200-µL reaction buffer (20 mmol/L citric acid, 60 mmol/L Na2HPO4, 1 mmol/L EDTA, 0.1% CHAPS and 1 mmol/L DTT, pH 6.0) for 30 min at 37 °C and analyzed by WB.
Cell apoptosis detection
The cell lines were treated with 50 μmol/L cisplatin for 12 h; (1–5) ×105 cells were collected and treated with BD Pharmingen™ FITC Annexin V Apoptosis Detection Kit (Cat#556547, BD Biosciences, New Jersey, USA) according to the manufacturer’s protocol. Samples were analyzed on a BD LSR Fortessa X-20 flow cytometer (BD Biosciences).
PAR-CLIP sequencing and data analysis
Stable U2OS cells transfected with pHY023-tG3BP1-C and pLKO.1-shAEP were cultured in 15 cm2 dishes. When the cell confluence reached 70%, 50 μmol/L cisplatin and 100 μmol/L 4-SU were added to the culture, and the cells were incubated for 12 h. The medium was aspirated, and cells were added with ice-cold phosphate-buffered saline (PBS). The lids were removed, and the cells were irradiated twice (150 mJ/cm2) in a UVP Crosslinker with a 365-nm lamp. The cells were harvested and lysed with an IP buffer (1 mmol/L DTT, 1 × complete protease inhibitor, RNase inhibitor added) for 20 min. The samples were centrifuged at 4 °C and 22 000 × g for 10 min, and the supernatant was collected carefully. RNase T1 (1 U/μL) was added to the lysate, which was incubated at 25 °C for 15 min. The lysate was added with prewashed protein A/G agarose beads (10 μL/100 µL) and an anti-Flag-tag antibody (1:100) and incubated overnight on a rocking shaker. On the following day, the beads were washed three times with a lysis buffer, and RNAs were extracted using TRIzol. RNA-sequencing was performed by Novogene.
To generate the count matrices, raw FASTQ files of PAR-CLIP sequencing data were processed using STAR, whereas the GRCh38/hg38 index genome was used for the alignment of the PAR-CLIP-seq data. PAR-CLIP-seq analysis was conducted using R (version 3.6.0) package Limma (version 3.56.0) to process the enrichment matrices. Gene Expression Omnibus and Kyoto Encyclopedia of Genes and Genomes enrichment analyses were performed with the online tool DAVID (https://david.ncifcrf.gov/tools.jsp), and the plots of these results were produced online by SangerBox (http://sangerbox.com/). The sequencing coverage histogram was generated with IVG (version 2.15.2).
DNA probes and reagents
All probes used in FISH were biotin-tagged and synthesized by Sangon Biotech Co., Ltd., and detail sequences are listed in Table S5. SA -onjugated Cy3 and FITC (Cat#D110512, Sangon Biotech Co., Ltd.) were used for FISH. Other reagents and buffers used in FISH were as follows: 20×SSC buffer, DEPC Treated (Cat# B548110, Sangon), Formamide deionized (Cat#A600211, Sangon).
FISH
U2OS cells were seeded in 12-well cell culture plates with round coverslips and treated with cisplatin (50 μmol/L) for 6 h. The cells were fixed with 4% formaldehyde at RT for 15 min. The FISH kit was purchased from GenePharma, and subsequent FISH assays were performed according to manufacturer’s protocol.
SUnSET assay
To monitor the protein synthesis of U2OS, 143B, and U87-MG stable cell lines after treatment with cisplatin, SUnSET assays were performed. These cell lines were seeded in 10-cm dishes and treated with 50 μmol/L cisplatin for 6 h until the confluence reach 80%. Puromycin was added with final concentration at 10 µg/mL and cultured for 1 h. The cells were collected, WB was performed, and anti-puromycin antibody was used for detection.
Polysome profiling with RNA-qPCR
To assess translational activity of target genes’ mRNA, cancer cell lines are treated with cycloheximide (CHX, 100 μg/mL, 5 min) to arrest ribosomes on mRNAs, followed by rapid harvesting and washing in ice-cold PBS containing CHX. Cells are lysed in a buffer containing 20 mmol/L Tris (pH 7.5), 150 mmol/L KCl, 5 mmol/L MgCl2, 1% Triton X-100, 1 mmol/L DTT, CHX, and RNase inhibitors to preserve ribosome-mRNA complexes. After centrifugation to remove debris, the lysate is layered onto a pre-chilled 10%–50% linear sucrose gradient (prepared in 20 mmol/L Tris, 150 mmol/L KCl, 5 mmol/L MgCl2) and subjected to ultracentrifugation (35 000 r/min, 4 °C, 2.5 h, SW41 rotor) to separate ribosomal fractions. Gradients are fractionated using a density gradient system with continuous UV monitoring, collecting 12–14 fractions corresponding to monosomes (80S) and polysomes. RNA is extracted from each fraction using TRIzol, treated with DNase I, and reverse-transcribed into cDNA. Target mRNA levels in polysome versus monosome fractions are quantified by qPCR with gene-specific primers, normalized to housekeeping genes total RNA input. Higher mRNA enrichment in polysomal fractions indicates active translation.
Protein synthesis assay with the Click-iT homopropargylglycine (HPG) system
U2OS, 143B, and U87-MG cells were synchronized using 200 nmol/L dexamethasone for 2 h. The medium was then replaced with dexamethasone-free DMEM. To prepare the Click-iT HPG working solution, a stock solution was diluted 1:1 000 in a prewarmed L-methionine-free medium to a final HPG concentration of 50 μmol/L. Thirty minutes before the designated time, the culture medium was replaced with an HPG working solution. After 30 min of incubation at 37 °C, the cells were washed once with PBS and fixed with 4% formaldehyde. Then, Alexa Fluor 488 azide was conjugated to HPG through a click reaction according to the manufacturer’s protocol. HPG-incorporated proteins were subsequently detected by fluorescence microscopy.
RNA IP (RIP)-qPCR assay
The RIP assay was conducted to validate the mRNA binding of RPS4X and MT-ND1 to tG3BP1-C. U2OS overexpressing flag-tagged tG3BP1-C1 and tG3BP1-C2 or control cells were cultured in 10-cm dish and treated with cisplatin (50 μmol/L) for 6 h. The cells were fixed with 4% formaldehyde at RT for 15 min, and the fixation was then stopped with 0.125 mol/L glycine (in PBS) for 5 min. The collected cells were washed twice with 1 mL of ice-cold PBS, spinned, and the supernatant was discarded. Moreover, 500 µL of the IP lysis buffer (protease inhibitor and RNase inhibitor included) was added and incubated on ice for 15 min. The lysate was centrifuged at 15 000 × g for 15 min at 4 °C, 50 µL (10% v/v) of the supernatant was taken and restored at 20 °C as input, 20 µL of the anti-flag antibody and isotype mouse IgG were added, respectively, and 50 µL Protein A/G Plus agarose beads were mixed to the rest lysate. The mixture was incubated on a rotating shaker at 4 °C overnight. The next day, the mixture was spinned at 4 °C, the supernatant was discarded, and beads were washed four times with ice-cold PBS. RNA was isolated from the input lysate and beads with RNAclean Kit (Cat#DP412, Tiangen Biotech) according to the manufacturer’s protocol. To quantify the mRNA level of RPS4X and MT-ND1, in input, IP, and IgG groups, RT-qPCR was performed. The specific primers for the above mRNA are listed in Table S5. The final data were analyzed according to the following formulas:
$$\begin\varDelta C_}=\left[_}-_}-_^}\right]\\ \varDelta C_/}=\varDelta C_}-\varDelta C_}\\ =^}_/}}\end$$
Ribosome profiling
To measure the translation of the target mRNA in NC and tG3BP1-Cs-overexpressed U2OS cells, ribosome profiling assay was performed. The steps for ribosome profiling were slightly modified based on the literature (Ingolia et al., 2012). RNAs in ribosome pellets extracted from the aforementioned procedure were purified with RNAclean Kit (Cat#DP412, Tiangen Biotech). The sequences of RT-qPCR primers are listed in Table S5.
RNA-motif pulldown assay
In the PAR-CLIP dataset, 10 candidate genes were identified, and the MEME suite was utilized (Bailey et al., 2015) for consensus motif prediction, ultimately selecting three candidate motifs. Subsequently, the motif sequences of two candidate genes were chosen, motifs were synthesized with approximately 30 nucleotides flanking RNAs and their respective antisense oligonucleotides, and 5’ biotin-modified RNAs were synthesized.
Separately, tG3BP1-C1/C2 was overexpressed in HEK 293 T cells in a 15-cm dish, and cell lysis was induced using RIPA cell lysis buffer to obtain the cell lysate including tG3BP1-Cs for the subsequent pulldown assay. Initially, the biotin-modified RNA was preincubated with streptavidin-magnet beads at RT for 1 h. Subsequently, the above cell lysate was added to each preincubated mixture and incubated overnight at 4 °C on a rocking shaker. The following day, the samples were washed four times with ice-cold PBS, and pulldown proteins were eluted using a 5× SDS sample loading buffer. Pulldown results were assessed by WB.
Detection of the mitochondrial membrane potential
The mitochondrial membrane potential was applied to estimate mitochondrial damage in stable cell lines. The stable cells were seeded in 6-well cell culture plates and treated with cisplatin (50 μmol/L) and appropriate vehicle solution for 6 h. The mitochondrial membrane potential assay kit with JC-1 (Cat# C2006, Beyotime) was used for the detection of mitochondrial membrane potential according to the manufacturer’s protocol.
Oxygen consumption rate (OCR) assay
Stable cell lines of U2OS, 143B, and U87-MG were seeded in 96-well cell culture plates and treated with cisplatin (50 μmol/L) or appropriate vehicle solution for 6 h. OCR Fluorometric Assay Kit (Cat# E-BC-F068, Elabscience, Wuhan, China) was used according to manufacturer’s protocol. In this experiment, JC-1 staining was used to assess mitochondrial membrane potential. The green signal represents JC-1 monomers (indicative of depolarized mitochondria, damaged mitochondria), while the red signal corresponds to JC-1 aggregates (indicative of polarized mitochondria, healthy damaged mitochondria).
Patient specimens
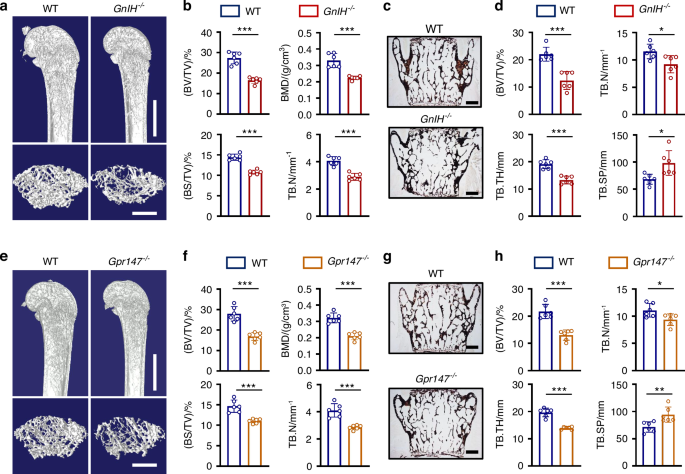
This study included 95 glioma and 36 OS specimens that were clinically and histopathologically diagnosed at Renji Hospital. Their diagnoses were independently rereviewed by two pathologists and classified by the World Health Organization criteria. Freshly frozen glioma tissues [low-grade glioma (LGG), n = 10; high-grade glioma (HGG), n = 72; recurrent glioma, n = 18] and five normal brain tissues were procured from the Neurosurgery Department of Renji Hospital. All the tissues were made into tissue chips.
In vivo xenograft model
Athymic male nu/nu mice (Lingchang Biotech, Shanghai, China) were used as glioma models. U87-MG cells (5 × 105/5 μL) were stereotactically injected into the right ganglia region. Chemotherapy treatment (cisplatin, 4 mg/kg BW) was initiated 7 days post tumor implantation to allow establishment of measurable lesions. Therapy was administered via intraperitoneal injection every 3 days for a total of 4 cycles. Mice were monitored daily and examined by magnetic resonance imaging (MRI) when they exhibited weight loss or neurologic impairment. Tumor diameters were measured from the MR images. The mice were euthanized when they were severely emaciated and depressed.
For the OS model, athymic female nu/nu mice aged 6 weeks were used. 143B cells (5 × 105/10 µL) were inoculated into the marrow cavity of the right tibia. After 20 days, the mice were sacrificed, and the tumors were isolated. The tumor volume was estimated by the diameter difference between ipsilateral and contralateral hind limbs.
For the in vivo assay, all chemical reagents were prepared in appropriate solution according to the manufacturer’s protocol. Their concentrations were indicated in our results. Treatment with chemical reagents began on day 7 after cell injection. Twelve tumor burden mice (glioma, n = 6; OS, n = 6) were treated with RR-11a (20 mg/kg BW, daily, i.p.). All the aforementioned mice and other mice xenografted with our stable transfected cell lines were treated with cisplatin (4 mg/kg BW, twice a week, i.p.), and negative control (NC) group was treated with normal saline solution.
Statistical analysis
Expression and survival analyses of patients with glioma and breast cancer were performed by Gene Expression Profiling Interactive Analysis (GEPIA), a web server for gene expression profiling in tumor and normal tissue and interactive analyses. Correlation analysis of the expression of two genes was also performed with GEPIA.
Statistical analyses were performed using IBM SPSS Statistics version 21.0 (IBM Corp., Armonk, NY, USA). Graphs were generated using GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA, USA). Two-tailed Student’s t test, one-way analysis of variance (ANOVA), Pearson correlation analysis, Kaplan‒Meier analysis, and log-rank tests were performed to analyze the corresponding data. A two-tailed P value < 0.05 was considered to indicate a significant difference. Grayscale analysis of WB bands and profiling and quantification of fluorescence intensity in fluorescence images were performed using Fiji (version 1.54 f).

Comments (0)