
Remember me
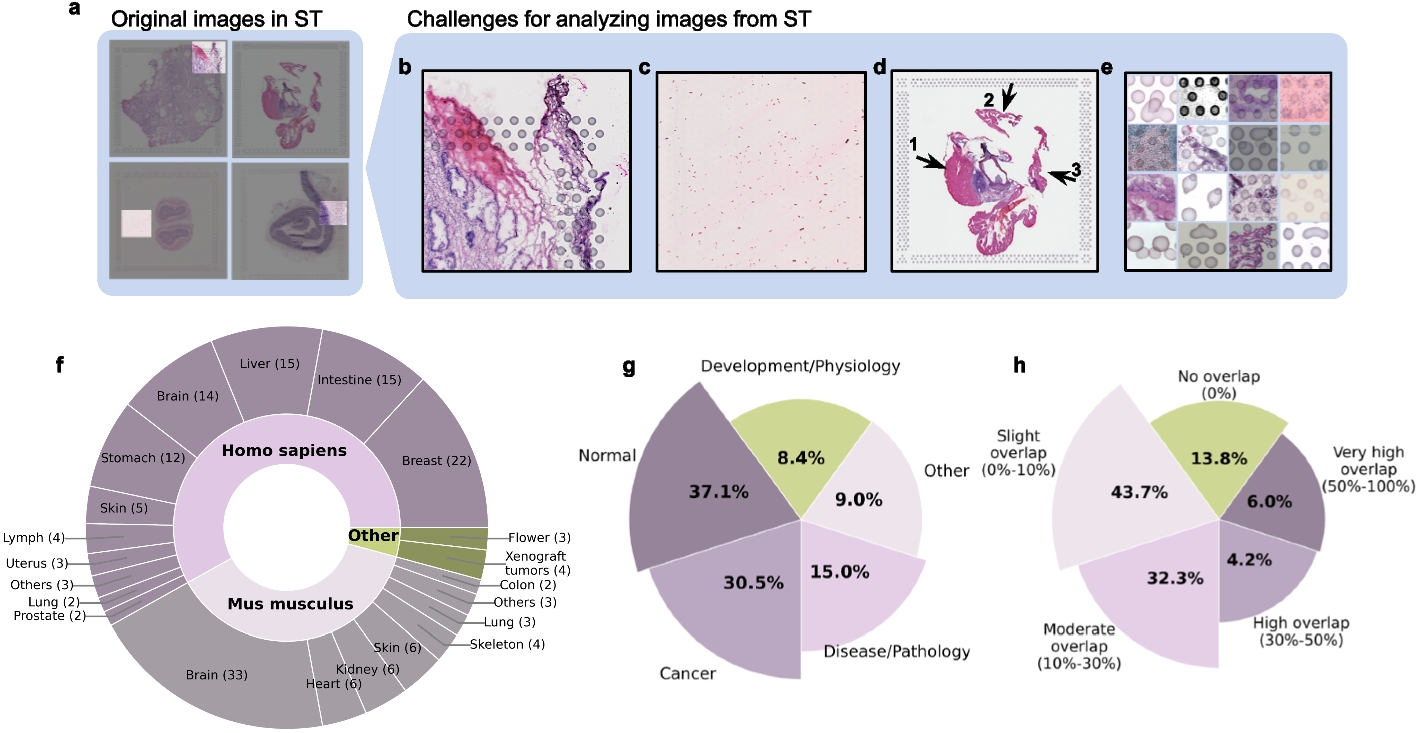
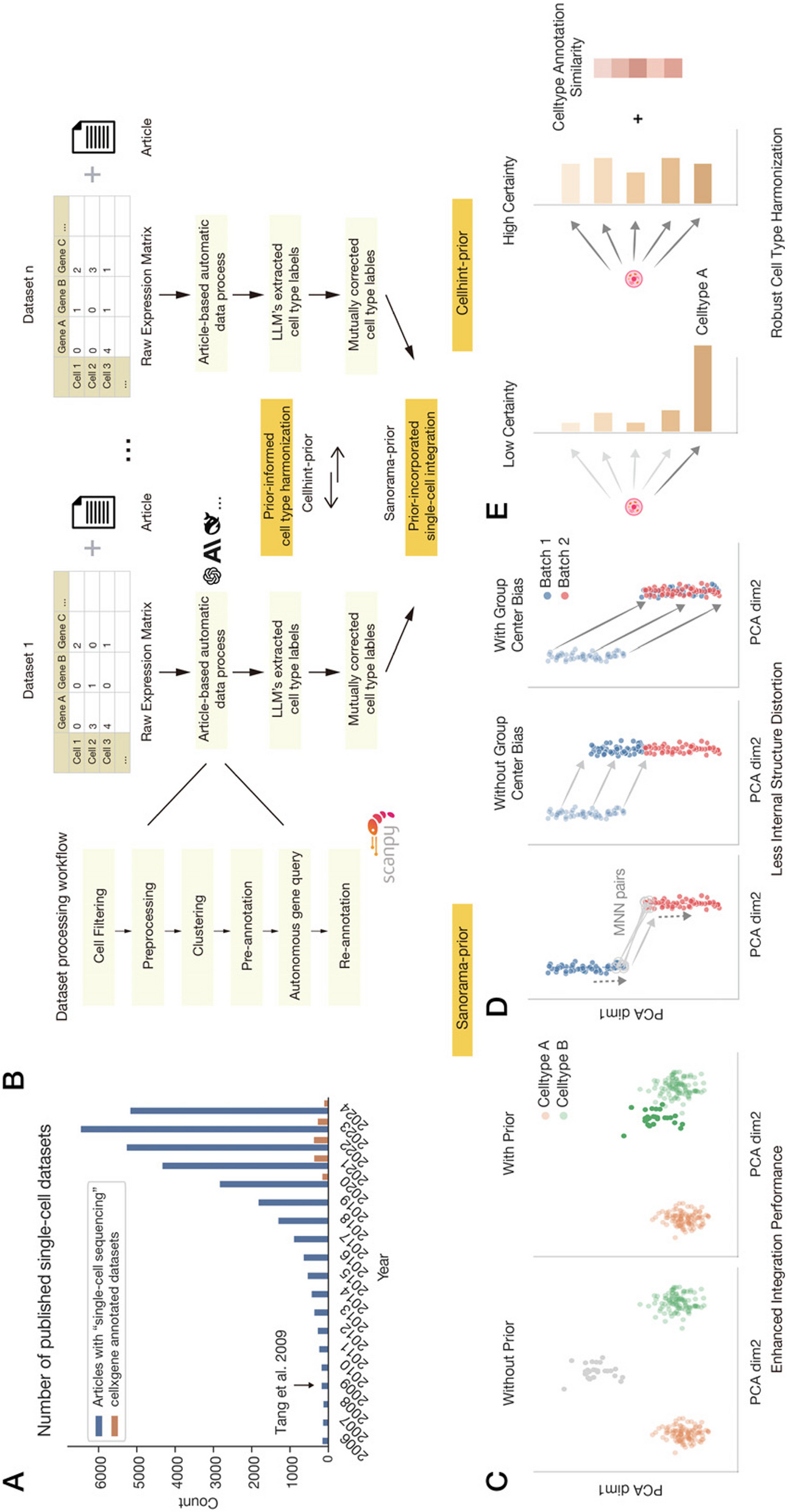


scExtract’s automated processing pipeline consists of two components: LLM-based automatic annotation incorporating article background information, and cell-type harmonization with embedding integration guided by annotation information (Fig. 1B). In the annotation phase, we implemented an LLM agent that emulates human expert analysis, automatically processing datasets while incorporating article background information. For the integration phase, to optimize the annotated data processing, we utilized the preliminary annotation information. We modified two well-performing software packages, scanorama and cellhint, and integrated them in a pipeline that leverages approximate annotations to enhance dataset integration.
In annotation stage, scExtract employs scanpy [10], the standard Python framework for single-cell data analysis, to perform computations. The standard processing pipeline includes cell filtering, preprocessing, unsupervised clustering, and cell population annotation. scExtract emulates the workflow adopted by human researchers in actual single-cell data annotation (Fig. 1B) (Methods), by extracting parameters used in each step from the article text and implementing them using the scanpy system. For instance, if the target article mentions in the “Methods” section, “We filtered out cells with ≥ 20% mitochondrial genes,” scExtract would extract this parameter and execute the corresponding computation.
In the clustering phase, scExtract’s prompts include two aspects: it can extract the number of cluster groups from the article as an external parameter, and when the article does not explicitly state the number of groups, it can infer from the article’s content, such as the number of cell populations discussed or the granularity of annotations. This method leverages the authors’ prior knowledge, which is crucial for preserving maximal biological significance in the clustering process. While algorithms exist for selecting optimal cluster numbers in unsupervised clustering, they are often not applied in practical data analysis due to their potential unreliability in capturing biologically meaningful distinctions.
During the annotation phase, similar to previous work utilizing LLMs for cell annotation, scExtract takes marker gene lists for each group as input. However, it also incorporates the article’s background knowledge, ensuring that annotation results align more closely with the article’s content. To better mitigate hallucinations in LLM information extraction and differences in the implementation platform of processing workflows, scExtract can optimize previous annotations after the initial annotation by autonomously querying the expression levels of a set of characteristic marker genes. This gene set is generated by scExtract based on the article and existing tissue and cell type information, inferring potential cell populations in the current microenvironment and characteristic genes of annotated but low-confidence groups. After obtaining the expression levels of these genes across different groups, scExtract can further optimize its previous annotations.
The next step in constructing a fully automated dataset is to integrate the data annotated by scExtract. However, the majority of conventional integration methods fail to leverage prior annotation information. To overcome these limitations, we developed scanorama-prior. Scanorama-prior requires additional clustering information and a similarity matrix of cell annotations at the embedding level, both from the scExtract processing pipeline. When constructing mutual nearest neighbors (MNN), scanorama-prior considers the prior differences between cell types (Fig. 1C), adjusting the weighted distances between cells across datasets, thereby achieving more accurate neighbor construction.
Additionally, when shifting cells between datasets, scanorama-prior tends to move original cell groups as cohesive units towards corresponding groups in the target dataset. To enhance batch correction, it applies an additional adjustment vector based on the positions of cell group centers in both datasets. The weight of this adjustment vector is determined by the annotation similarity between the original and target groups. Consequently, if two groups exhibit similar expression patterns and identical annotations, they will be more uniformly integrated (Fig. 1D). This approach ensures that biological relationships are preserved while effectively mitigating batch effects.
While possessing various advantages, scanorama-prior exhibits sensitivity to annotation errors, a characteristic we detailed in the benchmark section. To address this limitation, we incorporated cellhint-prior into scExtract’s integration method. Cellhint, originally designed for cluster-level integration, serves as an ideal downstream complement to rough annotations. Cellhint’s methodology can be employed to rectify previous annotations and provide harmonized annotations through cross-dataset comparisons. To mitigate potential interference from prior information, we adopted a conservative approach in incorporating prior knowledge by adjusting annotation weights based on cell alignment uncertainty levels (Fig. 1E)(Methods).
scExtract implements a stepwise integration process for automatically annotated datasets (Fig. 1B). The process begins with cellhint-prior for cell type harmonization, leveraging neighboring datasets as references to rectify potential nomenclature inconsistencies stemming from LLM output variations. When embedding-level integration is necessary, scanorama-prior is applied to the harmonized cell types. The text-to-embedding approach’s flexibility in cell type input format enables the similarity matrix to be derived from harmonized cell types. This pipeline effectively addresses both annotation harmonization and embedding integration challenges in heterogeneous single-cell omics datasets.
Evaluating clustering and annotation accuracy using cellxgene dataWe utilized manually annotated datasets from cellxgene to assess annotation accuracy. We randomly selected 21 medium-scale annotated datasets (on the order of 104 cells), 18 of which possessed diverse cell types (Fig. 2A, Additional file 2: Table S1) [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. These datasets encompass samples from multiple human tissues or organs, including liver, kidney, and intestine, addressing a wide range of distinct biological contexts. We compared scExtract with three established methods, including SingleR [28], scType [29], and CellTypist [30] (Methods). To ensure cost-effectiveness when applied to larger-scale data, we employed three model providers supportive of large-scale queries, with longer context (> 128 k tokens) and suitable pricing (≤ $5.00 per 1 M input tokens): Deepseek-v2.5 [31], GPT-4o-mini [32], and Claude-3.5-sonnet [33].
Fig. 2
scExtract automatically annotates published datasets with high accuracy. A Schematic workflow of the cellxgene datasets benchmark annotation task. Raw expression matrices from 18 cellxgene datasets were downloaded, processed through scExtract using three model provider APIs, and were benchmarked against curated annotations. B Dot plot showing the correlation between true cell types and automatically annotated cell types. Correlation coefficient calculated using Pearson’s correlation. C Bar plot showing the adjusted rand index between curated and annotated groups. D Dot plot illustrating the comparison of cell annotation similarity metrics on benchmark datasets, using text-to-embedding similarity. The left side shows the accuracy averaged at the single-cell level, while the right side shows the accuracy averaged at the group level. p-values calculated using Wilcoxon signed-rank test. E Box plot demonstrating the relationship between cell type annotation accuracy and data abundance across different cell populations. F Box plot comparing the improvement in annotation accuracy when incorporating literature knowledge versus direct annotation. p-values calculated using Wilcoxon signed-rank test. G Box plot showing the relationships between confidence levels assigned by scExtract using different LLMs and actual annotation accuracy. H,I Dot plot illustrating the comparison of cell annotation similarity metrics across four different annotation methods on benchmark datasets with Claude 3.5 sonnet as the annotation model. H shows text-to-embedding similarity measurements, while I presents cell-ontology similarity metrics. Within each panel, the left side displays cell-level average accuracy, while the right side shows cluster-level accuracy. p-values calculated using Wilcoxon signed-rank test. J UMAP plot displaying standard cell types in a kidney dataset (left) and cell types automatically annotated by scExtract using Claude 3.5 (right). K UMAP visualization demonstrating the text-to-embedding accuracy of cell type annotations generated by scExtract. L UMAP visualization showing cell type annotations generated by SingleR. M UMAP visualization illustrating the text-to-embedding accuracy of SingleR annotations. N UMAP visualization showing cell type annotations from a second independent run of scExtract using Claude 3.5 Sonnet, aligned with literature-defined cell types
We first evaluated the preservation of group structure in single-cell RNA-seq datasets across different automatic annotation methods. Regarding the discrepancy between annotated cell type numbers and those reported in the original articles, SingleR and CellTypist, which operate at the single-cell annotation level, exhibited a clear tendency to overestimate cell type numbers due to single-cell level noise. In contrast, scType and scExtract, which utilize marker gene lists for cell group annotation, demonstrated better correlation with curated cell type numbers (Fig. 2B). Among all model providers used in scExtract, Claude 3.5 tends to overestimate the number of cell types, while Deepseek-v2.5 and GPT-4o-mini tend to underestimate the number of cell types. We further assessed the clustering performance using the Adjusted Rand Index (ARI) (Fig. 2C). Group-level annotation methods demonstrated significantly higher structural similarity compared to single-cell-level approaches. Meanwhile, Claude 3.5, due to its propensity for detailed annotation, displayed the lowest ARI among all models.
To evaluate annotation accuracy, we employed large language model’s text-to-embedding method, converting the annotation information into embeddings and calculating cosine similarity (Methods). scExtract-based methods demonstrated higher mean accuracy than current established methods across all 18 datasets in text-to-embedding (Fig. 2D). Moreover, reference transfer methods are more likely to accurately capture common cell types comprising the majority of the dataset due to their frequent occurrence in reference datasets. We observed that the annotation accuracy of all methods decreased as the cell proportion in the overall dataset declined (Fig. 2E). To address this limitation of single-cell level average, we implemented a cluster-level accuracy metric that disregards the influence of cell numbers. This metric better reflects the accuracy of automatic annotation for rare cell types. Using this metric, scExtract methods showed a clear advantage (Fig. 2D). The performance differences within various language models is less pronounced, with Claude 3.5 demonstrating the most accurate information extraction capability, followed closely by Deepseek v2.5, and then GPT-4o-mini, which is roughly consistent with these models’ performance in other domains. Additionally, we also evaluated the relationships between standard and annotated cell types using Cell-Ontology label (Methods). The results were consistent with embedding-based methods, with a more pronounced disparity between group-level and single-cell-level approaches in reference transfer methods (Additional file 1: Fig. S1A).
Compared to the previously published GPTCelltype [7] method, we hypothesize that scExtract’s superior annotation accuracy stems from its application of article-derived prior knowledge and self-reflective capability through two-round annotation. During the annotating step, scExtract completed its annotations based on background and characteristic genes, then reviewed and adjusted the annotation content. For instance, in the annotation of actual datasets, during the second round of annotation, scExtract subdivided intercalated cells into two subgroups by querying the expression levels of SLC26A7 and SLC26A4 in the second round (Additional file 1: Fig. S1B-D).
We evaluated the performance difference between scExtract with and without background knowledge integration (Fig. 2F). Methods incorporating background knowledge demonstrated improved accuracy, and we hypothesize that the varying degrees of improvement among different models depend on their ability to capture contextual information from the background text. We also observed that as text was proportionally replaced with confounding contents, annotation accuracy declined to varying degrees, although more sophisticated models like Claude3.5 exhibited a comparatively smaller decrease in performance (Additional file 1: Fig. S2A-B).
We directly compared the accuracy of GPTCelltype and scExtract methods on our dataset across different models and metrics, with scExtract consistently demonstrating significant improvements than GPTCelltype (Fig. 2H,I, Additional file 1: Fig. S2C-D). When assessing annotation accuracy using text-to-embedding methodology, the performance hierarchy was clearly established: scExtract > scExtract (no context) > GPTCelltype. Similarly, in cell ontology-based accuracy measurements, the performance ranking was as follows: scExtract > scExtract (no context) ≈ GPTCelltype. Notably, the performance differences between scExtract (no context) and GPTCelltype varied according to the specific accuracy metric employed. We attribute these variations to inherent methodological differences in the evaluation approaches. The text-to-embedding method may be more susceptible to formatting inconsistencies, particularly regarding standardized Cell Ontology annotation usage, whereas the Ontology Look Service (ols_api) effectively neutralized these differences by systematically mapping annotations to the standardized Cell Ontology hierarchical structure.
To mitigate the black-box nature of LLMs, we also implemented confidence scoring during the annotation process (Fig. 2G). Results validated that the confidence scores provided by scExtract reliably correlate with actual annotation accuracy.
scExtract accurately annotated datasets with accordant group structureTaking the scExtract-generated dataset of adult human kidney [15] annotated by Claude 3.5 as a representative example (Fig. 2J). The structure largely aligns with the clustering in the original article, demonstrating accuracy in annotation granularity. This contrasts with reference transfer-based methods, which often misclassified single cell types into heterogeneous populations due to expression fluctuations (Fig. 2L). Regarding annotation accuracy, most cell clusters were annotated consistently with the original definitions.
However, a small portion of cells showed lower accuracy (Fig. 2K), typically author-defined cell subtypes more susceptible to language model fluctuations. For instance cells annotated as “Unknown” by scExtract (Fig. 2J) were originally defined as PT_VCAM1 [15] (subpopulation of proximal tubule with VCAM1 expression). Interestingly, this subtype was correctly annotated as “Proximal tubule (PT_VCAM1)” in the second replicates, matched to the original article (Fig. 2N, Additional file 1: Fig. S1D). Among model providers, Claude 3.5’s annotations adhered more closely to the original text, while Deepseek v2.5 and GPT-4o-mini tended to reduce the specificity of original expressions, providing more generic annotations (Additional file 1: Fig. S3A). These observations underscore the necessity for further refinement and validation of LLM-based annotations.
We also evaluated the poorer-performing neural tube dataset (Fig. 2D, Additional file 2: Table S1 dataset 4). Results indicated that scExtract’s lower performance largely stemmed from overly broad cell type definitions in the standard dataset, with more accurate cell type annotations in the original article (Additional file 1: Fig. S3B). For example, scExtract’s automatic annotations of two import structures, roof and floor plate cells, aligned well with marker gene expression patterns (Additional file 1: Fig. S3D). However, as clustering complexity and customization increased, annotations required more careful consideration.
Overall, scExtract maintained granularity and annotation fidelity while operating fully automatically. It can process novel datasets cost-effectively without relying on pre-annotated reference datasets, offering significant advantages in scalability and extensibility. Notably, scExtract can complete all process procedure of a dataset in less than 20 min (Additional file 1: Fig. S3E) at a cost of less than one dollar, with no additional computational resources required if using web API. Moreover, with the ongoing advancements in language models, we anticipate further improvements in precision and efficiency.
Evaluating scExtract’s output stability and prompt sensitivityMajor constraints limiting the applications of large language models in scientific domains include output variability and susceptibility to prompt interference. We next assessed the performance of the LLM agent in scExtract with respect to these aspects. We selected Sample1, Sample11, and Sample19 (Additional file 2: Table S1), repeating the analysis of each sample for six times to evaluate performance across two task phases: parameter extraction during preprocessing and cell annotation labeling.
For preprocessing parameters, high consistency was observed when parameters were explicitly stated in the manuscripts (Additional file 1: Fig. S4A-B). Variability occurred primarily for non-critical parameters not explicitly defined, where the LLM made reasoned inferences and cautiously set them to “default” rather than fabricating values. Reduced reproducibility was noted in complex scenarios such as manuscripts containing multiple datasets with different characteristics, unpublished manuscripts, or custom algorithms (Additional file 1: Fig. S4C). For cell annotations, high reproducibility was achieved for most cell types across replicates. Variability was typically confined to cell subpopulations with ambiguous marker profiles (Additional file 1: Fig. S5A). Solid tissue samples (Samples 11, 19) (Additional file 1: Fig. S7A-B) showed higher annotation consistency than blood samples (Sample 1) (Additional file 1: Fig. S5A). Annotation variability correlated with cell population heterogeneities and was accurately reflected in the model’s self-reported certainty levels (Additional file 1: Fig. S7A, S8B). When comparing different methods, we observed that strict reproducibility ranked as GPTCelltype > scExtract (no context) > scExtract (Additional file 1: Fig. S5A-B, S6A). However, this ranking inversely correlated with annotation specificity, as more detailed annotations naturally introduced more potential variations. Methods lacking article context failed to identify novel cell subtypes discussed in the original papers (Additional file 1: Fig. S9A-B).
We tested prompt sensitivity using Sample 11 with five prompt variants: instruction sequence reordering, terminology modification with synonyms, enhanced guidance with detailed instructions, minimalistic annotation with minimal instructions, and structural reformatting using JSON output format (Methods). Results showed that parameter extraction was generally robust across prompt variations. Claude 3.5 Sonnet maintained more consistent performance with simplified prompts compared to Deepseek v3 (Additional file 1: Fig. S10A-B). Similarly, annotation content remained largely stable across prompt variants, particularly for well-defined cell types (Additional file 1: Fig. S11A-B) (Additional file 3: Table S2). Advanced models demonstrated reduced sensitivity to prompt variations (Additional file 1: Fig. S11A-B).
In conclusion, scExtract maintains robust performance across multiple replicates and prompt variations, particularly for well-defined parameters and cell types. Variability occurs primarily in scenarios requiring nuanced interpretations of ambiguous data. The implementation of certainty reporting and detailed reasoning logs provides users with transparency regarding the model’s decision-making process, enabling appropriate interpretation of the results.
Scanorama-prior enhanced integration of annotated single-cell omics datasetsWe evaluated scanorama-prior’s integration performance using pancreas single-cell RNA-seq datasets, which utilized different sequencing platforms on pancreas samples [34]. In contrast to the original version’s integration results, scanorama-prior demonstrated superior batch effect removal while preserving cell type differences (Fig. 3A). Utilizing identical resolution parameters, unsupervised clustering on the adjusted embeddings revealed that scanorama’s results displayed more intra-cluster separations resembling batch distributions, primarily due to incomplete batch effect removal. We also benchmarked corrected PCA and UMAP embeddings on batch correction and biological diversity preservation [6]. Results showed that scanorama-prior’s embeddings in both spaces exhibited superior performance metrics compared to the original version (Fig. 3B).
Fig. 3
Prior incorporated integration method benchmark of human pancreas dataset. A UMAP plot comparing the integration performance of Scanorama (upper panels) and Scanorama-prior (lower panels) on pancreatic datasets. Each column from left to right shows batch, cell type, and the results of unsupervised clustering using the integrated embedding. B Table presenting the batch correction and biological conservation performance of Scanorama, Scanorama-prior, and unintegrated datasets. Overall aggregate results are displayed in the horizontal barplot on the right. C,D Tree plots illustrating the performance of the original Cellhint (C) and Cellhint-prior (D) on pancreatic data. Each column represents a batch, with dots representing cell type groups within the batch. Horizontal lines between dots indicate they were aligned across datasets. E UMAP plot demonstrating the integration of pancreatic data in UMAP dimensions using Cellhint (upper panels) and Cellhint-prior (lower panels)
To evaluate scanorama-prior’s robustness to incorrect labeling, we tested its integration performance under three scenarios: similar naming (cell types replaced with aliases or alternative nomenclature), unbiased incorrect naming (erroneous naming without bias towards existing cell types, e.g., annotated as unknown), and biased incorrect naming (erroneous naming biased towards existing cell types, e.g., mislabeling one cell type as another). Our findings revealed that scanorama-prior performed well in the first two scenarios. In the first case, the integration efficacy was essentially indistinguishable from that achieved with strictly identical string inputs (Additional file 1: Fig. S12A). In the second scenario, the method adopted a more conservative approach to integrate the two cell types, yet still managed to merge them based on expression patterns (Additional file 1: Fig. S12B). However, in the third scenario, scanorama-prior received conflicting directives from two dimensions. Consequently, it refrained from integrating the cells into either group, instead positioning them as outliers between the two clusters (Additional file 1: Fig. S12C). This observation underscores the importance of ensuring basic similarity in annotation types when integrating datasets, or at the very least, avoiding biased incorrect annotations.
Cellhint-prior enhanced cross-dataset annotation harmonization with improved tolerance for naming errorsTo address this challenge in fully automate context, we leveraged a prior-aware version of cellhint, which can automatically reconcile annotation discrepancies across datasets. Evaluation using pancreas datasets revealed that while the original cellhint excelled in integrating inDrop series data, it struggled with Smart-seq2 and Fluidigm C1 platforms (Fig. 3C). In contrast, the prior-informed cellhint demonstrated superior alignment compared to its predecessor. Notably, it successfully aligned cells with severe batch effects due to disparities in batch and dataset origins (Fig. 3D, E). This improvement underscores the value of incorporating annotation priors in the cell type alignment process, particularly when dealing with heterogeneous data sources. We further evaluated cellhint-prior’s performance under those three previously mentioned annotation error scenarios. Results demonstrated that the structure of the cell type harmonization tree remained consistent with that of correct annotations across all three conditions (Additional file 1: Fig. S12F). This stability underscores the method’s enhanced robustness to annotation variations.
scExtract’s two-step automated integration showed robust performance on mis-annotated cell typesRegarding the previously mentioned annotation error tolerance, we conducted multiple experiments to evaluate the impact of incorrect annotations on the scExtract two-step integration pipeline (Methods). In each experiment, we implemented all three types of label changes and isolated the modified data for subsequent benchmark evaluation (Additional file 1: Fig. S12A-C). Our assessment of integration performance metrics revealed that, consistent with previous benchmark results, scanorama-prior with prior knowledge performed optimally before label changes, while the use of cellhint for harmonization showed no significant impact (Additional file 1: Fig.S13A). However, after label changes, scanorama-prior without cellhint-prior harmonization showed marked performance degradation, while the scExtract’s prior-harmonized version maintained robust performance (Additional file 1:Fig.S13A).
Additionally, we performed unsupervised clustering on the post-change embeddings and annotated each cell cluster using majority voting. In scenarios where label changes resulted in cells being separately clustered, as in previous experiments, these cells would be misclassified. We observed that scanorama-prior-harmonized demonstrated the highest tolerance to annotation errors (Additional file 1: Fig. S13B-F), in some cases surpassing the tolerance of cellhint raw without prior knowledge integration. This pattern was also evident at the embedding level (Additional file 1: Fig.S14A-E). Scanorama-prior, obtained through our two-step integration approach, demonstrates optimal merging between cells with altered type labels (dark labels) and other cells of the same type (light background). This superior performance is attributed to two key factors: enhanced batch effect correction, compared to cellhint, and improved resilience to annotation variations, compared to un-harmonized scanorama-prior.
Comprehensive evaluation of large-scale atlas integration demonstrates the scalable superiority of scExtract-based methodsWe evaluated time performance of our two-step automated integration method on concatenated large datasets (Methods). scanorama-prior, which incorporates a prior similarity matrix, demonstrated slightly higher time complexity compared to the original scanorama method (Additional file 1: Fig. S15A) in a PC-level processer. To overcome the computational limitations of scanorama-based methods on large datasets, we implemented GPU acceleration. When executed on a V100 GPU, scanorama-prior exhibited speed increase compared to the original no-prior method and demonstrated scalability to datasets with one million cells (Additional file 1: Fig. S15B). Notably, in our benchmark tests, cellhint-based methods exhibited significantly longer execution time compared to those reported in the original paper [9] (Additional file 1: Fig. S15B). This discrepancy arises from our approach of generating large datasets by concatenating smaller ones, where the original dataset size increases proportionally with cell numbers, which is more reasonable in a real scenario. In contrast, benchmark in the original study employed downsampling from large datasets, maintaining constant original dataset numbers. We observed substantial runtime differences between these two approaches. However, since our raw datasets were relatively small, this represents an extreme case demonstration. In practice, actual execution times are expected to fall between these two extremes.
We further evaluated scExtract’s integration performance on six larger datasets [6, 9, 35], containing between 50,000 and 600,000 cells in total, representing typical real-world integration scenarios. We first tested the impact of adding general cell type descriptions to the integration process. Interestingly, we found virtually no differences in similarity matrices generated using either “cell type” alone or “cell type: description” (Additional file 1: Fig. S16A-B). This suggests that standard cell type designations already serve as sufficient sources of prior information. As cellhint’s integration approach modifies cell adjacency relationships rather than directly altering embeddings in reduced dimensional space, we generated 2D UMAP embeddings using identical parameters for benchmarking. In datasets around 50,000 cells level (Fig. 4A, Additional file 1: Fig. S17A), scanorama-prior, derived from scExtract’s two-step pipeline, demonstrated superior batch correction and biological preservation compared to both the original scanorama and other methods. This was followed by scanorama itself, with cellhint-prior and cellhint showing relatively poor performance. While we anticipated that direct embedding displacement would be more effective than graph relationship modification for smaller datasets, we hypothesize that single-cell noise might compromise the effectiveness of such approaches in larger-scale datasets.
Fig. 4
Assessment of scExtract’s two-step integration strategy on large-scale datasets. A–C Systematic evaluation of integration performance on Immune cells (A), Blood cells (B), and Human Lung Cell Atlas (C) datasets. Dataset statistics (number of cells, datasets, and cell types) are indicated above each panel. Upper panel: UMAP visualizations of embeddings generated by different methods (from left to right): PCA, cellhint integration, cellhint_prior integration, original scanorama, and scExtract’s two-step integration derived scanorama_prior. Lower panel: Performance metrics (from left to right): scatterplot showing batch effect removal versus biological variation preservation, barplot of comprehensive performance metrics, barplots displaying KMeans Adjusted Rand Index (ARI), Silhouette label scores, integration Local Inverse Simpson’s Index (iLISI), and k-nearest neighbor Batch Effect Test (kBET)
For datasets containing more than 200,000 cells, we observed substantial variations in method performance across different datasets. In the Blood and Spleen datasets, containing 200,000 and 330,000 cells respectively but only four datasets each, benchmark results indicated that cellhint-prior and cellhint significantly outperformed other methods, with minimal performance differences between them (Fig. 4B, S17B). The original scanorama showed relatively poor performance, consistent with benchmark results reported in the corresponding literature. However, in the Human Oral Cell Atlas and Human Lung Cell Atlas examples, which comprised 250,000 cells/14 datasets and 580,000 cells/14 datasets, scanorama-prior, derived from our two-step integration, regained the leading position, while prior-knowledge-enhanced methods demonstrated increasingly larger advantages over their original counterparts (Fig. 4C, S17C).
Based on these results, we hypothesize that high-cell-count, low-dataset-number configurations may compromise the performance of embedding-based methods while favoring graph-construction approaches. From a practical perspective, we recommend terminating the integration process at the cellhint-prior step when the average cell number per dataset exceeds 50,000 or when dealing with fewer than four datasets, as this approach may yield optimal results. However, this modification does not diminish the significance of prior annotation, as both cellhint and cellhint-prior require pre-clustered data input.
Additionally, for novel datasets without reference materials, we tested first using scExtract (no context) to annotate the datasets, and then integrating them using scExtract’s two-step approach. We conducted analyses using pancreatic datasets as examples and performed three replicates. The overall performance of scExtract (no context) fell between that of scanorama-prior using standard cell types and the original scanorama (Additional file 1: Fig. S18). These results are reasonable from our perspective. When examining specific performance aspects, the batch correction effect of scExtract (no context) integration was significantly better than the original scanorama, approaching the level of scanorama-prior that directly used standard cell types for integration. However, in terms of biological preservation, scExtract (no context) showed a decline, likely due to potential annotation errors interfering with the integration steps. Notably, across different replicates, the composite scores remained highly robust (Additional file 1: Fig. S18), consistent with our previous conclusions from the annotation evaluation section.
Automated construction of a skin autoimmune disease dataset with custom data incorporationThe skin serves as a critical barrier for our body, comprising various cell types. It is crucial for skin health to maintain homeostasis within its microenvironment [36]. Autoimmune conditions like psoriasis and atopic dermatitis (AD) often result from dysregulated micro-environmental signaling pathways involving complex interactions among keratinocytes, immune cells, fibroblasts, and other cellular components [37, 38]. Despite recently emerging large-scale datasets [39, 40], they often encompass limited disease phenotypes and require substantial resources for de novo single-cell sequencing. We utilized scExtract to automatically integrate skin datasets, addressing these limitations.
We screened 20 articles for dataset construction (Additional file 4: Table S3), excluding six due to species mismatch or inconvenient accession of raw data. Phenotype labels were manually curated from NCBI information, with acceleration of scExtract by extracting data accession and metadata of samples. Standard automated integration procedures using Claude 3.5 yielded a comprehensive dataset of 440,000 cells from the remaining 14 articles (Fig. 5A, B) [41,42,43,44,45,46,47,48,49,50,51,52,53,
Comments (0)