
Remember me
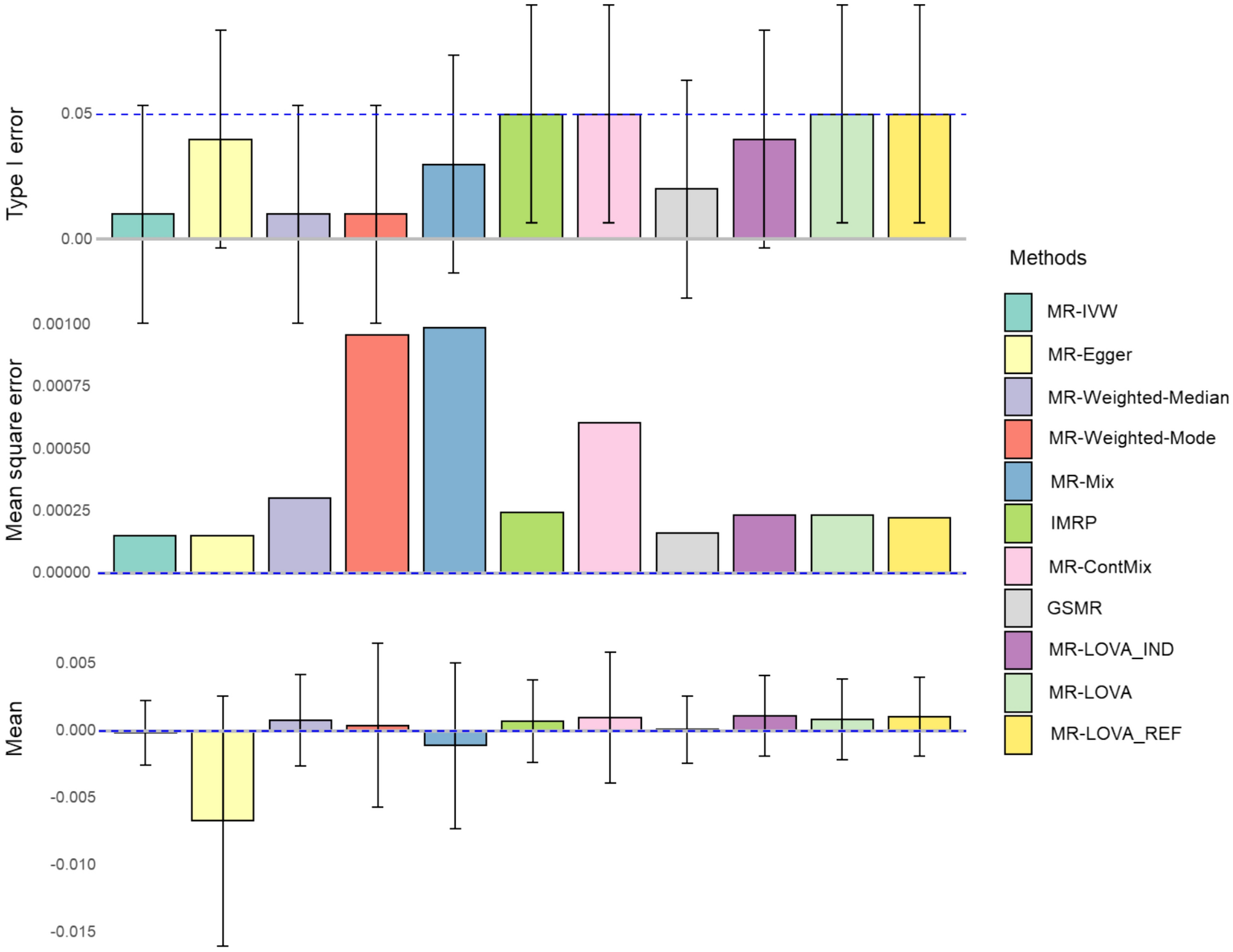
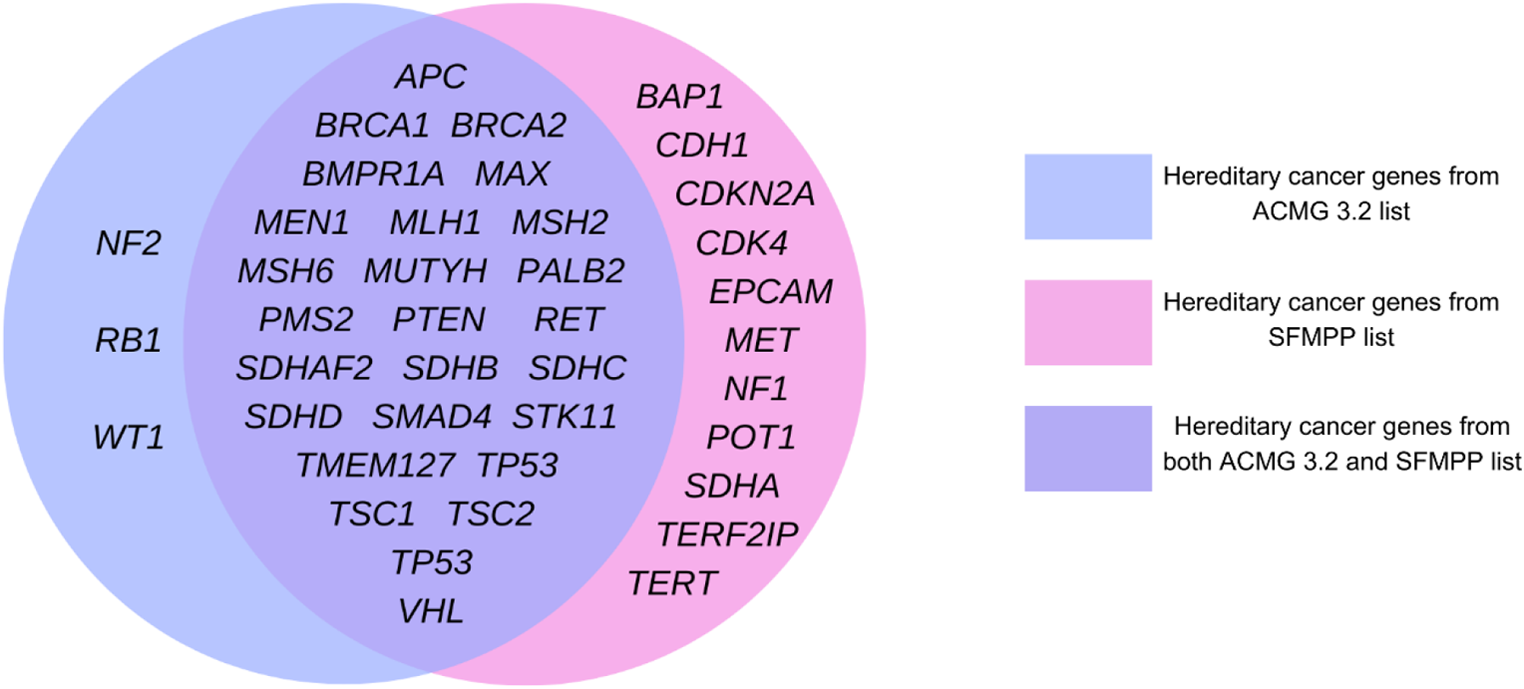



The clinical variant model (CVM) was developed, consisting of two binary classification steps. First, the patient score model distinguishes clinical presentations consistent with condition(s) associated with particular genes, and those that are not. Second, the variant score model synthesizes the evidence from multiple patients with a given variant to infer the probability of pathogenicity (Fig. 1a).
Fig. 1
Overview of clinical variant model. a CVM stepwise approach to leveraging clinical data to predict the pathogenicity of variants for a given genetic condition. b The patient score: clinical-related patient information is gathered for all patients with a molecular diagnosis for the condition as well as for all patients who have only benign variation in the gene of interest and no other molecular diagnosis (genotype-negative cohort). A two-part machine learning pipeline is trained to distinguish the individuals with molecular diagnosis from genotype-negative individuals. The trained model is then applied to each individual in the entire cohort to generate a patient score. c The variant score: Each stack represents a known benign or pathogenic variant in a gene of interest. Each box within a stack represents a patient. Patients with low patient scores are shown in white, and patients with high patient scores are shown in black. A Bayesian inference model infers a variant score for each variant based on its associated patient scores
Study population and internal datasetsPrivacy and ethicsThe Western Independent Review Board protocol number CR-001-02 (Tracking ID 20161796) approved the use of de-identified patient data for all analyses.
Clinical cohortClinical and genotype data were obtained for individuals referred by clinicians between January 2015 and April 2024 to a diagnostic laboratory for germline genetic testing. Patient descriptors (“clinical data”) about each individual were extracted from the test requisition form (TRF), including age at testing, sex, race, ethnicity, and ancestry (REA), specialty of the ordering provider, ICD-10 codes, and unstructured text included the indication for testing and family history. Additional preprocessing steps are described in Supplementary Methods. Genotype data for each individual included genes tested and all variants identified by testing.
ConditionsA CVM condition was defined as a set of genes and their associated modes of inheritance (MOIs) for a molecular condition. Molecular conditions and associated annotations, such as the condition name, were drawn from the unique Mondo Disease Ontology (Mondo) (Shefcek et al. 2020) identifiers which had at least one strong or definitive gene association curated by the Gene Curation Coalition (GenCC) database (DiStefano et al. 2022). For each Mondo entity, multiple CVM conditions could be created by including subordinate disease entities within the Mondo ontology, potentially yielding multiple distinct CVM conditions. Additional conditions were added by request from expert scientists when they were not automatically generated. For each condition, a dataset was assembled consisting of clinical data for patients (probands or relatives), genotypes for the included genes, and existing variant classifications within those genes.
Patient and variant labelsThe patient score and variant score steps each required known (“labeled”) examples– of patients and variants, respectively.
For the patient score model, patients were categorized as molecular diagnosis, control, or indeterminate. The molecular diagnosis cohort was identified according to logic based on the gene-inheritance associations for each condition. For example, for a monogenic condition with autosomal dominant inheritance, the molecular diagnosis cohort included patients with at least one pathogenic (P) or likely pathogenic (LP) variant in the gene. The control cohort consisted of patients who had only benign (B) or likely benign (LB) variants observed in any associated gene. Patients were considered indeterminate if they met criteria for neither labeled cohort. For each condition, 20% of labeled patients were randomly held out as a test set for model evaluation, and another 20% was used as a validation set for training assessment, consistent throughout model training.
For the variant score model, variants were classified by Sherloc (Nykamp et al. 2017), a numeric point-based adaptation of the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al. 2015), and assigned to one of three categories– pathogenic (including both P and LP variants), benign (B and LB) or VUS. A stratified 20% sample of labeled variants per gene was held out as a test set and excluded from parameter estimation.
Patient score modelA transformer-based large clinical language model (Gatortron-S, UNFLP) (Yang et al. 2022), pre-trained on MIMIC-III notes, PubMed, and other sources, was fine-tuned using curriculum learning, which performed better than direct transfer learning (Supplementary Methods). First, the language model was trained on pooled and deduplicated patient examples from across all 1,334 conditions. The model was further fine-tuned to each condition individually (Supplementary Methods). Predictions from the fine-tuned language model were then combined with other clinical data, to train a second model (XGBoost) (Chen et al. 2016), which produced the final patient score (Fig. 1b).
Bellwether patientsFor each variant, a cohort of “bellwether” patients was identified—patients whose genotypes and gene-inheritance pairs made their observations valuable for classifying the variant. Under the assumption of complete penetrance, a bellwether patient would manifest the disease if the variant were pathogenic. Decision trees for identifying bellwether patients in single-gene autosomal recessive and autosomal dominant models are shown in Supplementary Fig. 1, with logic for other MOIs and multi-gene models shown in Supplementary Table 1.
Variant score modelFor inferring variant pathogenicity, a custom Bayesian hierarchical probabilistic graphical model (PGM) was developed and implemented with Pyro (Bingham et al. 2018). Variables, their prior distributions and likelihood functions, were determined according to clinical genetics knowledge. This setup implies a full joint probability distribution and defines the predictive distribution.
The variant score model used partial pooling to jointly infer gene-level penetrance (fraction of bellwether patients for pathogenic variants expected to appear affected with disease), phenocopy (fraction of bellwether patients for benign variants expected to appear affected with disease) and pathogenic rates (fraction of included variants in a gene that are pathogenic) for multiple genes. Stochastic variational inference was used to fit the model to patient scores and variant labels for both labeled variants and VUS for the training set for each condition. The variant score was sampled from the posterior predictive distribution by conditioning the inferred model on the patient scores for a given variant (Fig. 1c).
Model validation, post-processing, and selectionFirst, overall performance for each model was assessed according to discrimination, classification and calibration metrics measured over held out samples. Only models which demonstrated high discrimination performance, as measured by the area under the receiver operating characteristic curve (AUROC of ≥ 0.8) on both steps of the pipeline were nominated for further selection. At the variant score level, posterior probability thresholds were applied to variant scores to identify variants receiving high-confidence benign or pathogenic predictions corresponding to pre-specified positive and negative predictive value targets (PPV ≥ 0.99; NPV ≥ 0.95). Observed predictive value performance was then estimated on the held out set to ensure generalization of these posterior thresholds. Additional filters were applied at the variant level. Variants with fewer than eight bellwether observations were excluded from receiving confident benign predictions; fewer than three for pathogenic. Additionally for pathogenic predictions, two of the three bellwether patients were required to be unrelated and have patient scores greater than 0.5.
Models and variants were further filtered by an independent team of scientists according to potential impact of this data on variant reclassification and operational constraints. The team also independently evaluated the suitability of patients for inclusion in the model, the concordance of patients’ presentations with the resulting patient scores, and the confidence and directionality of the variant-level predictions for each high-confidence prediction.
Incorporation into a variant classification frameworkFor models and individual predictions that passed all validation criteria, clinical evidence was used to award points in Sherloc. Variants with predictions corresponding to ≥ 0.95 NPV received 3 B points, sufficient to classify the variant as LB without evidence to the contrary, while those with ≥ 0.99 PPV were awarded 3 P points, insufficient to reach a LP classification without additional corroborative evidence.
Comments (0)