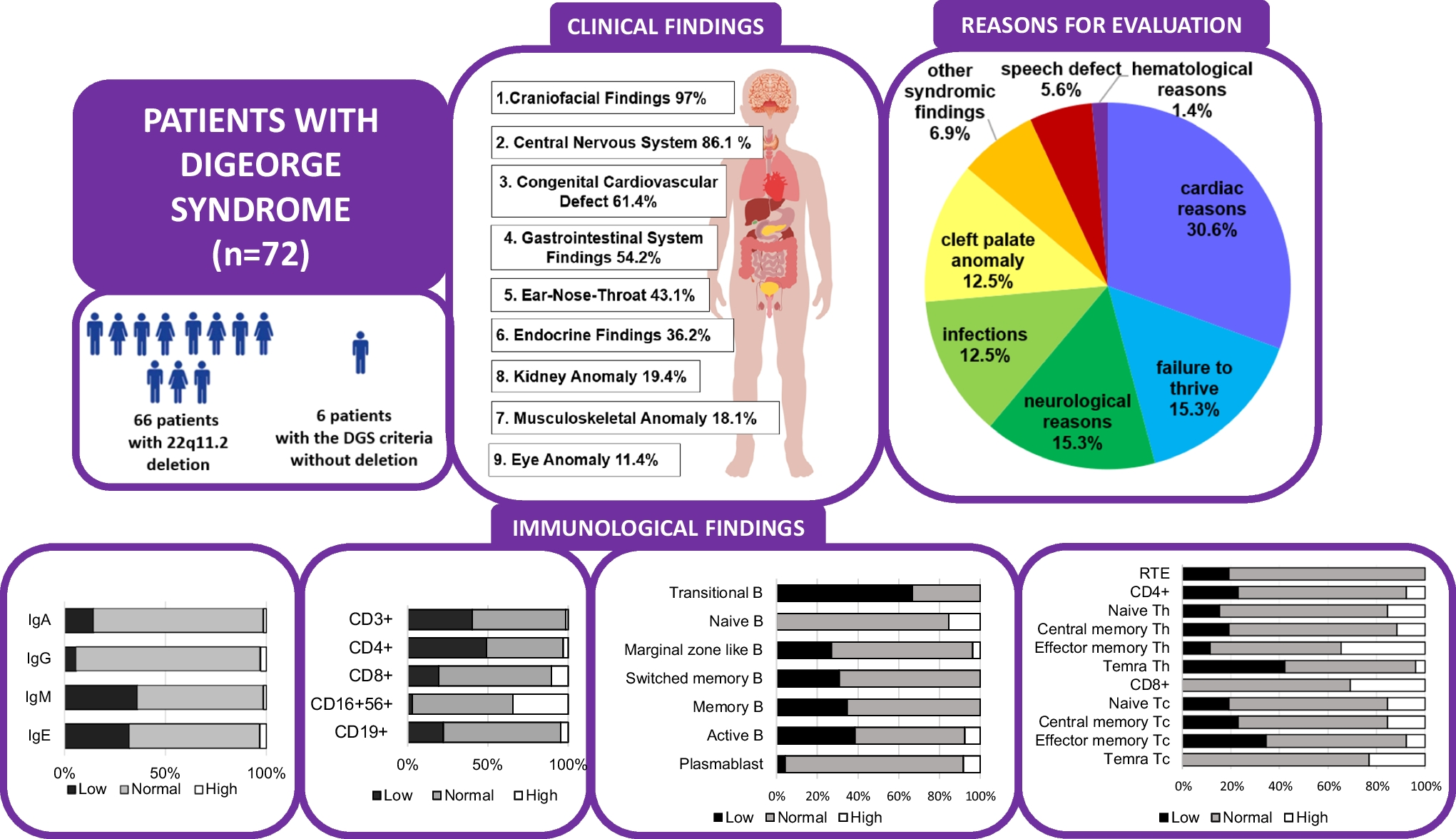
Study Subject
The case is an African-American man enrolled in the Multicenter AIDS Cohort (MACS), a study of HIV infection in men who have sex with men ongoing in 4 US sites since 1984 [19]. MACS participants have semiannual study visits at which medical and behavioral history are recorded, physical exam and laboratory testing are performed, and plasma, serum, and viable peripheral blood mononuclear cells (PBMC) are frozen [19]. In another study, cryopreserved PBMC from this man surprisingly did not express CD16 by flow cytometry. Therefore, further studies were undertaken.
At enrollment in the MACS in 2003, the case was 42 years old, was receiving combination antiretroviral therapy (cART), and was virologically suppressed (plasma HIV RNA < 50 copies/ml by Roche ultrasensitive assay (Roche Diagnostics, Nutley, NJ)), with normal white blood count, hemoglobin A1c concentration, and serum concentration of alkaline phosphatase (Table 1). Later, when the MACS performed more extensive testing, he had slightly elevated creatinine, alkaline phosphatase, and gamma-glutamyl transferase in serum and total protein in urine but did not show any signs of immunodeficiency or persistent infections. He had cleared infection with hepatitis B virus and no infection with hepatitis C virus. His HIV-related history (Table 2) showed undetectable plasma HIV RNA at most study visits, with CD4 T cell counts generally > 500/µL when he was virologically suppressed. Intermittent non-suppression of HIV replication was possibly due to reduced adherence to antiretroviral therapy (Table 2). At both study entry and at follow-up study visits he reported no hospitalizations or other illnesses.
Table 1 Laboratory data of the caseTable 2 HIV-related laboratory data of the caseAfter identification of his CD16 deficiency, when he was 61 years old, a more detailed medical and family history was obtained, and serum concentrations of total complement, complement components C3C and C4C, IgG and its subclasses, IgM, and IgA were measured in stored serum at Quest Diagnostics (Frederick, MD). He did not recall having had any unusual, serious, or persistent infections, had no history of recurrent warts or herpetic lesions, had never been told of any problems with his immune system, and had no family history of infections or early death, including both parents and a half-sister who was alive and healthy. Serum concentrations of complement and immunoglobulins were normal, except that concentration of IgG1 was slightly higher than the normal range (Table 2).
Flow Cytometric Analysis of CD16A Expression
CD16A expression was analyzed in PBMC from (a) the case, (b) a healthy HIV-uninfected donor described previously [21], and (c) a MACS participant who is living with HIV and had similar age and percentage of NK cells among peripheral lymphocytes as the case. NK cells (CD45+CD3−CD56+/CD16+) and monocytes (CD45+CD3−HLA-DR+CD14+/−) [22] in PBMC were identified with monoclonal antibodies (mAbs, from BD Biosciences (San Jose, CA) unless otherwise noted): anti-CD14-brilliant violet (BV) 605, anti-HLA-DR-BV650, anti-CD3-fluorescein isothiocyanate (FITC), anti-CD56/CD16-phycoerythrin (PE), anti-CD45-peridinin-chlorophyll-protein (PerCP), and Near-IR LIVE/DEAD™ Fixable dye (Invitrogen, Carlsbad, CA). Surface expression of CD16 was assessed with two mAbs: anti-CD16-BV421 (clone: B73.1) and anti-CD16-BV510 (clone: 3G8), which identify different epitopes on CD16 [5, 23]. To assess intracellular expression of CD16A, cells stained with all mAbs above except anti-CD16A, fixed and permeabilized with Cytofix/Cytoperm™ (BD Biosciences), and stained with anti-CD16A BV421 and anti-CD16A-BV510. Data were acquired on a LSRII cytometer (BD Biosciences) and analyzed using FlowJo 10.7.1 (FlowJo, Ashland, OR). Gates were set using fluorescence-minus-one (FMO) controls [24], as shown in Fig. S1. PBMC from the case were initially tested in three experiments, using cryopreserved PBMC from 2 study visits 4.5 years apart. Later, we also analyzed CD16A expression on cryopreserved PBMC obtained from the case at his earliest available MACS study visit.
The following mAbs (all from BD Biosciences) were used to assess CD16A expression in NK cells and monocytes: anti-CD3-brilliant ultra violet (BUV)805, anti-CD56-BUV615, anti-CD16 -BV421 (clone: 3G8), anti-CD19-FITC, anti-HLA-DR-PE-Cy7, anti-CD14-Allophycocyanin (APC), anti-CD16-R718 (clone: B73.1), and Yellow LIVE/DEAD™ Fixable dye (Invitrogen). Data were acquired on a FACSymphony™ A5 cytometer (BD Biosciences) and analyzed using FlowJo 10.10.0 (FlowJo).
Genetic Analyses of FCGR3A
Although locus-specific PCR and Sanger sequencing suggested a mutation in the FCGR3A and FCGR3B loci, because of the high homology between these two genes we chose to use 10X Genomics Chromium™ (10X Genomics, Pleasanton, CA) linked-read sequencing to obtain phased parental reads that would span both loci [25, 26].
High molecular weight genomic DNA (50 kilobase (kb)– 1 + megabase (Mb)) was isolated from thawed PBMC of the case using the Nanobind CBB Big DNA Kit (Circulomics, Baltimore, MD) per manufacturer’s instructions, quantified using a Qubit fluorimeter, and diluted to 1.25 ng/ul in Tris-EDTA (TE) buffer. The DNA was processed using the Chromium platform (10X Genomics) and a sequencing library prepared following the manufacturer’s protocol [27]. The library was quantified both by qPCR and by Bioanalyzer (Agilent, Santa Clara, CA) electrophoresis and sequenced on a NovaSeq 6000 (Illumina, San Diego, CA) to generate 855.8 million reads with a mean read length of 139 bp (bp) after trimming and a mean read depth of 36.7X. Longranger™ v.2.2.2 (10X Genomics) was used to phase reads by their shared index. The average molecule length was 67.5 kb, and the N50 phase block size was 14.26 Mb.
The case’s haplotypes were compared to the NA12878 reference genome [28] available from 10X Genomics. Separate deletion events were predicted on each parental chromosome (Fig. 2a) with a retained FCGR3A-3B fusion allele. We designed PCR primers flanking the retained allele to narrow the cross-over position and performed Sanger sequencing on the products (Fig. 2b). All of the PCR, 10x Genomics, and both types of sequencing were carried out at the JHU Genetic Resources Core Facility, RRID: SCR_018669.
Immunophenotyping of NK Cells
Cryopreserved PBMC from the case and 5 other MACS participants matched with the case by HIV status, study visit, age (± 5 years), and percentage of total NK cells (identified as CD45+CD3−CD16+/CD56+) among lymphocytes were thawed and stained with the antibodies listed in supplementary Table 1. In experiments 1 and 2, all antibodies were combined in a single panel; however, this panel did not resolve NKG2C well, so in experiment 3 antibodies were divided into 2 panels and anti-NKG2C-PE was used. Each experiment included PBMC from the case (from the same study visit in experiments 1 and 2 and a study visit 4.5 years later in experiment 3) and from two or three matched donors. After surface staining, cells were fixed and permeabilized with eBioscience™ Foxp3 / Transcription Factor Staining Buffer Set (ThermoFisher Scientific, Waltham, MA) per manufacturer’s instructions and stained intracellularly with anti-FcRγ-FITC, anti-TCF-1-PE, and anti-(TBX-21)-PE-Cy7. Data were acquired on a FACSymphony™ A3 cytometer (BD Biosciences) and analyzed using FlowJo 10.7.1 (FlowJo). Gates were set using fluorescence-minus-one controls [24]. Total NK cells were identified as live/dead−CD3−CD4−CD8−CD14−CD19−TBX-21+ cells [29]. All NK markers were examined on total NK cells and on CD56bright, CD56dim, and CD56neg subsets of NK cells to account for differential expression of these markers with differential CD56 expression [30]. In experiment 3 Uniform Manifold Approximation and Projection (UMAP) was used for dimensionality reduction [27], and Flowsom to identify NK cell subsets based on marker expression [31].
Measurement of NK Cell FunctionSpontaneous Cytotoxicity and ADCC
Spontaneous cytotoxicity and ADCC of NK cells were assessed in three experiments, each using PBMC from the case (from one study visit) and a matched control. Target cells were K562 cells (American Type Culture Collection (ATCC)-CCL-243, Manassas, VA) for spontaneous cytotoxicity and Raji cells (ATCC-CCL-86) for ADCC. Both cytotoxicities were assessed as described [32,33,34], except that target cells were labelled with CellTrace™ Violet (Invitrogen) instead of Cell Tracker Orange. Briefly, target cells were labeled with CellTrace™ Violet as described [21]. Labeled Raji cells were incubated with either 10 µg/mL rituximab (generously donated by Genentech, South San Francisco, CA), or human IgG1 (Sigma Aldrich, St. Louis, MO) as a negative control, for one hour at 37 °C. PBMC from the case and matched controls were then mixed in triplicate with target cells at effector: target (E: T) ratios of 20:1, 10:1, 5:1, 2.5:1, and 0:1. After incubation for 4 h at 37 °C, cells were labeled with 1 µg/mL 7-aminoactinomycin D (7-AAD, ThermoFisher Scientific) for 20 min to allow discrimination of dead cells, then analyzed on a FACSCanto II cytometer (BD Biosciences) with dead target cells identified as 7-AAD+ CellTrace™Violet+. Percent cytotoxicity was then calculated as follows:
% spontaneous cytotoxicity= (% dead K562 cells with PBMC added \(\:-\:\)% dead K562 cells without PBMC added)/(100 \(\:-\:\)% dead K562 cells without PBMC added)
%ADCC= (% dead Raji cells with PBMC added to rituximab-coated targets \(\:-\)% dead Raji cells with PBMC added to IgG1-coated targets)/(100 \(\:-\:\)% dead Raji cells with PBMC added to IgG1-coated targets)
Cytokine Production
NK cells were purified from cryopreserved PBMC by magnetic bead negative selection using a Human NK Cell Isolation Kit (Miltenyi Biotec, Gaithersburg, MD). The phenotypic and functional effects of cytokine treatment of NK cells were assessed as previously described [35, 36]. Briefly, NK cells were seeded in 96-well round-bottom plates at 1 × 105 cells per well, and cultured for 48 h in serum-free AIM V medium (ThermoFisher Scientific) alone, or with addition of interleukin (IL)-18 (1 µg/ml; MBL International, Woburn, MA), or IL-18 in combination with IL-2 (1000 IU/ml; Proleukin; Prometheus Laboratories, San Diego, CA) or with IL-15 (200; R&D Systems, Minneapolis, MN). Cells were stained with mAbs to IFN-γ-AF700 (clone B27, BD Biosciences), MIP-1β-APC (clone D21-1351, BD Biosciences), CD3-APC-H7 (clone SK7, BD Biosciences), CD56-PC7(clone N901, Beckman Coulter, Indianapolis, Indiana) and with Aqua LIVE/DEAD™ Fixable dye (Invitrogen), and then production of IFN-γ and MIP-1β by NK cells was assessed by flow cytometric intracellular cytokine staining as described above for intracellular staining of CD16A. Secretions of IFN-γ and TNF-α into the culture supernatant were assessed by multiplex electrochemiluminescence immunoassay (Meso Scale Diagnostics, Rockville, MD).
Immunophenotyping of Monocytes and DC
Cryopreserved PBMC from the case and 3 matched controls were thawed and stained with the antibodies listed in supplementary Table 2. Sequential staining of CX3CR1, CCR3, CXCR2, CD14 and CCR2 at 37 °C was performed as described [37] to ensure optimal staining of chemokine receptors on monocytes. Data were acquired on a FACSymphony™ A5 cytometer (BD Biosciences) and analyzed using FlowJo 10.10.0 (FlowJo). Gates were set using FMO controls [24]. Because of the lack of CD16A expression on monocytes of the case, an alternative strategy to identify classical, intermediate, and nonclassical monocytes based on expression of CD14 and CD89 was developed and validated (see details in Supplementary Material).
Functional Analysis of Monocytes
Monocytes were isolated from cryopreserved PBMC by CD14 positive magnetic beads (Miltenyi Biotec) according to manufacturer’s specifications, and were cultured in Iscove’s Modified Dulbecco’s Media (IMDM; Gibco, ThermoFisher Scientific) containing 10% fetal bovine serum and 0.5% gentamicin (Gibco) in a 24-well flat bottom plate at 5 × 105 cells/well. Following a 24-hour culture in either media alone; IFN-γ (1000 U/mL; R&D Systems) and TLR4 agonist LPS (1ng/ml; Sigma); or IFN-γ (1000 U/mL; R&D Systems) and TLR7 agonist CL307 (750ng/ml; InvivoGen, San Diego, CA), secretions of IL-10, TNF-α, IL-6, and IL-1β into the culture supernatants were measured by multiplex electrochemiluminescence immunoassay (Meso Scale Diagnostics).
Generation and Functional Analysis of Monocyte-Derived DCs
Monocyte-derived DCs were generated as previously described [38]. Briefly, immature DC were generated by culturing monocytes isolated as described above for 5 days in Iscove’s Modified Dulbecco’s Media (IMDM; Gibco) containing 10% fetal bovine serum and 0.5% gentamicin (Gibco) in the presence of granular-macrophage colony-stimulating factor (GM-CSF) (1000 IU/mL; Sanofi-aventis, Bridgewater, NJ) and IL-4 (1000 IU/mL; R&D Systems). High IL-12p70-producing mature DC were generated by an additional 48 h of culture in the presence of a cocktail of maturation factors containing IFN-α (1000 U/mL; Schering Corporation, Kenilworth, NJ), IFN-γ (1000 U/mL; R&D Systems), IL-1β (10 ng/mL; R&D Systems), TNF-α (25 ng/mL; R&D Systems), and poly-IC (20 ng/mL; Sigma).
Phenotypes of immature and mature monocyte-derived DCs were assessed by staining the cells with anti-CD83-PE (clone HB15e, Beckman Coulter), anti-CD86-PE (clone FUN-1, BD Biosciences), or anti-Siglec-1-PE (clone 7-239, BD Biosciences), followed by data acquisition on a LSRFortessa cytometer (BD Biosciences) and analysis using FlowJo 10.10.0 (FlowJo). Mature DCs were exposed to CD40L-transfected J558 cells (J558-CD40L; a gift from Dr. P. Lane, Birmingham, UK) to induce their production of IL-12p70 as previously described [39]. Briefly, the DC were plated (2.5 × 104 cells/well) in a 96-well flat-bottom plate and stimulated with J558-CD40L (5 × 104 cells/well) for 24 h. Culture supernatants were collected and tested for concentrations of IL-12p70, IL-10, IL-6, IL-8, and TNF-α by multiplex electrochemiluminescence immunoassay (Meso Scale Diagnostics).
Testing for Antibodies to Herpesviruses
Antibodies to EBV, CMV, herpes simplex virus (HSV)-1/2, varicella-zoster virus (VSV), human herpesvirus (HHV)-6, HHV-7, and HHV-8 were assayed in stored serum using enzyme-linked immunosorbent assays (ELISA) at the National Cancer Institute (Frederick, MD) for antibodies to EBV, CMV, HHV-6 and HHV-8 [40] and at Quest Diagnostics for antibodies to HSV-1/2, VSV, and HHV-7.
Nucleic Acid Extraction and Quantification of Herpesviruses
Nucleic acid was isolated using a NucliSENS EasyMAG automated extraction system (BioMérieux, Boston, MA) from 1 ml stored plasma spiked with a fixed amount of DNA internal control virus (a complete DNA genome of the phocine (seal) herpes virus type 1, PhHV) [41], and subjected to quantitative real-time PCR targeting EBV, CMV, HHV-6, HHV-8, and PhHV as described [40, 42].
Statistical Analyses
Differences in spontaneous cytotoxicity and ADCC between the case and the control MACS participants were compared using multilevel mixed effect models, accounting for intra-person and intra-experiment correlations. The non-parametric Mann–Whitney U Test was used to compare expression of markers for NK cells, monocyte and DCs between the case and control donors. Analyses were performed using Stata version 15.0 (StataCorp, College Station, TX). A p value of < 0.05 was considered statistically significant.





Comments (0)