2.1 Lack of Natural History Data and/or International Classification of Diseases Codes
In general, RDs often have poorly understood disease pathophysiology, large variations in disease manifestations, and high unpredictability [24, 25]. Furthermore, a dearth of International Classification of Diseases, Tenth Revision diagnosis codes and natural history cohorts makes it difficult to characterize RDs, understand real-world treatment patterns, and establish a standard of care. Without the relevant data, this information is often derived from descriptions within case reports, physician interviews, and, if available, electronic medical records. The paucity of natural history data and International Classification of Diseases codes poses several issues for HTAs. It hinders accurate estimation of the size of the treatment-eligible population, adding substantial uncertainty to a budget impact assessment. Long-term extrapolation of treatment effect is difficult without reliable natural history data, potentially impacting survival and quality-adjusted life-year estimates and, ultimately, the outcome of a cost-effectiveness assessment for an orphan drug.
The lack of established treatments compounds the problem. Rare disease clinicians and patients are left to trial-and-error approaches [26], resulting in a highly variable care pathway. From the HTA perspective, this results in substantial variability between various pieces of (often scarce) evidence and complicates the selection of appropriate comparators. For example, among 78 research questions for 20 orphan drugs undergoing a full HTA in Germany between 2011 and 2021, active comparators were available for 58% of the research questions, with the remaining assessments using best supportive care or watchful waiting only as a comparator [27]. This poses an issue for both cost-effectiveness and budget impact assessments, as the cost of supportive care or symptomatic-only treatment is often relatively low compared with the costs of active treatment, particularly with novel agents.
2.2 Small Sample Sizes
In RDs, a clinical trial with 100–150 patients can be considered large. Within such a trial, randomization may be 2:1, meaning that one third of patients receive placebo or, if available, an active comparator. Subgroup analyses may contain cohorts of < 30 patients, which often necessitate non-parametric statistical tests. As an example, in a sickle cell disease gene therapy trial, only 25 of 35 total patients could be evaluated for an event rate analysis [28]. Even if trial results appear compelling, small numbers can prevent the detection of statistically significant differences and can lead to a judgment that the results are inconclusive. Symptom heterogeneity in RDs can magnify these difficulties. Measuring a symptom benefit may require the assessment of several endpoints in one trial, compounding the challenge of adequate statistical power in small trial populations.
As the trial data often directly inform economic modelling, challenges in demonstrating a treatment effect because of the small sample size are likely to translate into uncertainty around survival and quality-adjusted life-year estimates in the model. This issue is particularly relevant for subgroups, which may include very few patients in an RD trial.
2.3 Use of Non-Traditional Study Designs
Health technology assessment agencies have made it clear that they consider randomized clinical trials to provide the highest level of evidence on the efficacy and safety of a drug [29,30,31]. While we agree, it should be noted that Ethics Review Boards often reject the use of a placebo arm for severe RDs, especially those that are rapidly progressing or that lack approved treatments. Although effect size estimations may be calculated using a naturalistic or pre-specified historical control (external control arm), these techniques can be viewed as less valid compared with randomized clinical trials. Furthermore, generation of comparative evidence via a network meta-analysis is often impossible because of the paucity of studies and heterogeneity of populations and trial designs. This often necessitates the use of alternative indirect comparison methodologies, such as simulated treatment comparisons or matching-adjusted indirect comparisons [32]. The latter method is often more practical for use in HTAs that list few comparators but require an assessment of multiple outcomes of interest. However, in the setting of a limited sample size in many orphan drug trials, the effective sample sizes in a matching-adjusted indirect comparison may be very low and the resulting evidence may be deemed inconclusive and inappropriate to inform decision making.
2.4 Lack of Robust and Disease-Specific Study Endpoints
A positive regulatory decision is uncertain for most orphan drugs in development, partly because of disagreement on trial endpoints. If established endpoints exist in an RD, they often “borrow” assessments from more common diseases, even though these insufficiently capture symptoms or treatment benefit in RDs. For example, physicians treating neuromyelitis optica spectrum disorder typically have a background in multiple sclerosis, where the Expanded Disability Status Scale [33] is commonly employed to measure disability. However, the Expanded Disability Status Scale fails to capture the full range of vision disability in neuromyelitis optica spectrum disorder, as vision is only one of six parameters that it assesses. This makes it more difficult to measure treatment benefit in neuromyelitis optica spectrum disorder trials using the Expanded Disability Status Scale.
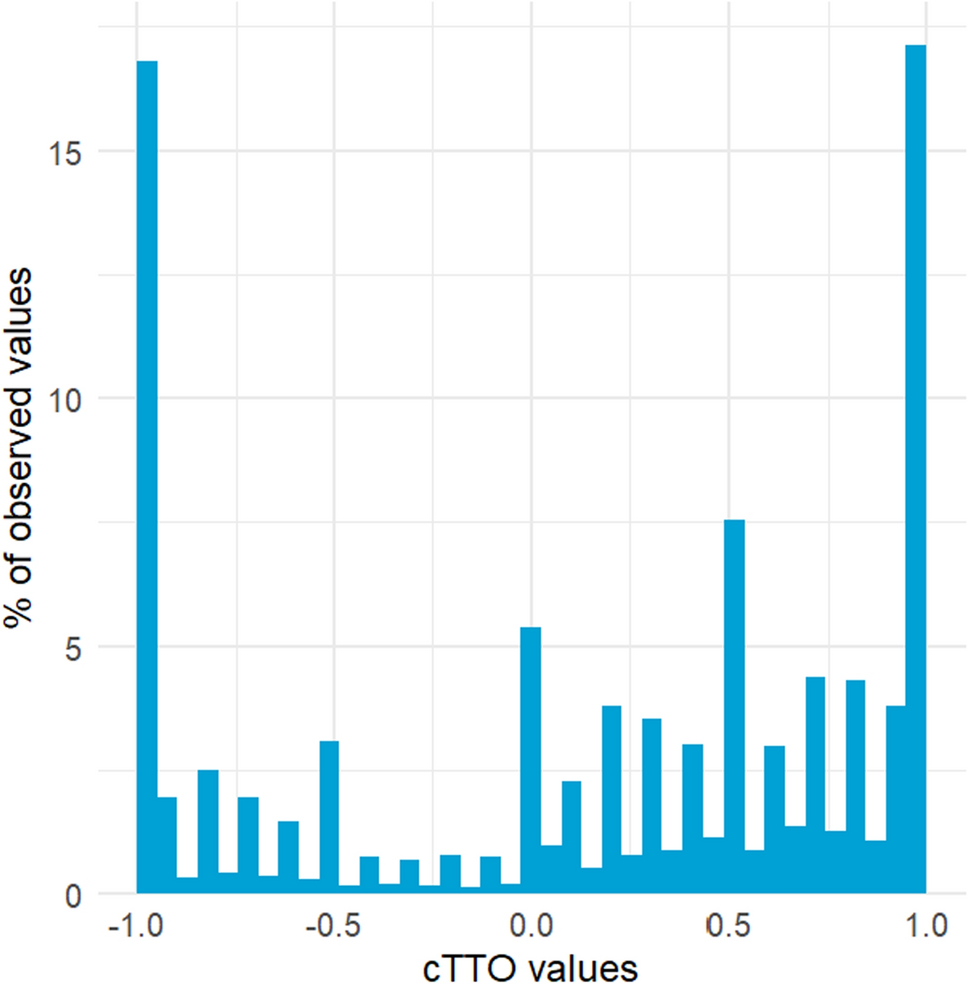
Furthermore, disease-specific patient-reported outcome measures do not exist for most RDs, so trials commonly employ generic instruments such as the 36-item Short Form Survey [34,35,36]. The lack of patient-reported outcome measures that adequately capture outcomes important to patients with RDs and their caregivers might lead to an underestimation of (or the complete omission of) the effects of an intervention. This issue emerged for example during the recent NICE appraisal of a gene therapy for transfusion-dependent β-thalassemia, where concerns were raised about the ability of the non-disease specific EQ-5D questionnaire to adequately capture the symptom burden and treatment effect [37].
While the generation of new endpoints in an RD is a possible solution, it takes years to establish and validate an endpoint and, because of the small size of the affected population, validation of the robustness of a novel endpoint in an RD is particularly challenging. An example of such challenges is well illustrated by the pivotal trial of an enzyme replacement therapy for mucopolysaccharidosis type VII, an ultra-RD with a global prevalence of less than 1 in 1 million individuals [38]. The trial, which enrolled 12 patients, assessed clinical efficacy based on the Multi-Domain Responder Index, combining several independent, clinically relevant assessments [38]. However, because of the heterogeneity of the disease presentations (including its cognitive impact), not all components of this endpoint could be completed by all patients, resulting in a substantial proportion of missing data [38]. Consequently, the regulatory assessment for this orphan drug primarily considered only a single component of the Multi-Domain Responder Index, the 6-Minute Walk Test [39, 40], which is a non-disease specific measure of walking capacity and endurance, commonly used in clinical trials across multiple therapeutic areas.
Another approach used in RD trials is to combine a disease-specific measure and a broad generic measure into a composite endpoint, as exemplified by a recent trial in Rett syndrome, which combined the Rett Syndrome Behaviour Questionnaire and the Clinical Global Impression-Improvement scale into a composite co-primary endpoint [41, 42]. However, this is not a one-size-fits-all solution, as it requires the availability of a disease-specific endpoint with some established validity (such as the Rett Syndrome Behaviour Questionnaire [43]), which may not be an option for ultra-orphan diseases or scenarios in which the regulatory pathway, where endpoint selection plays a key role, is highly uncertain.
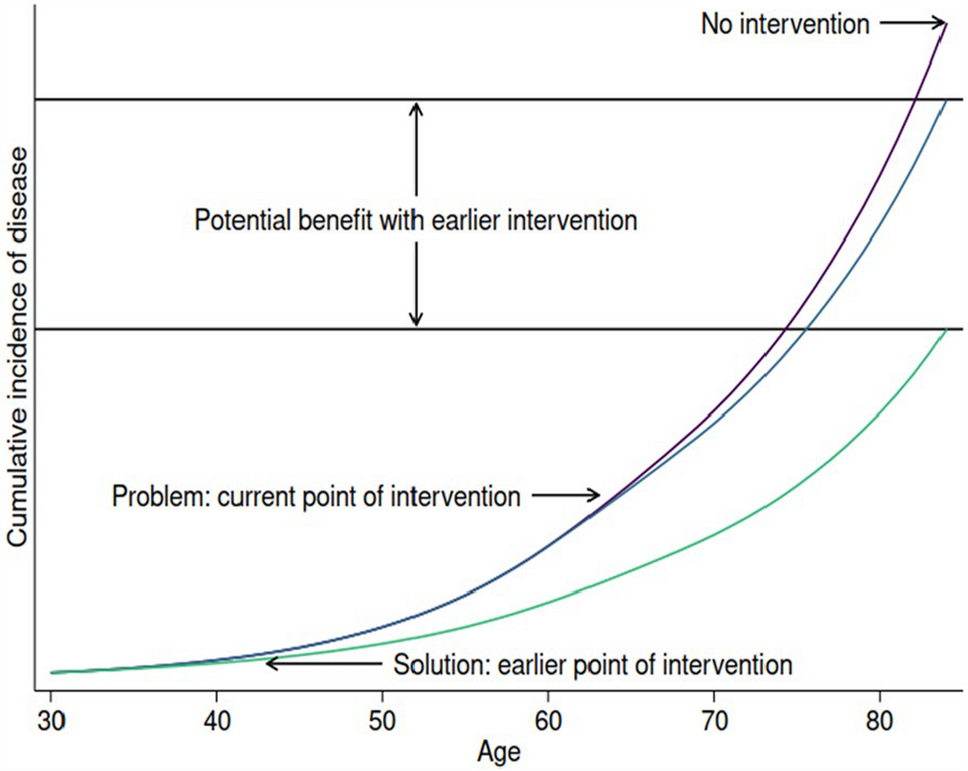
Time is also of the essence when considering endpoint development in RDs. Many RDs have no approved therapies and, as patients with RDs desperately await treatment options, manufacturers usually face a trade-off between demonstrating the ability of an intervention to mitigate disease progression over the shorter term versus potentially longer drug development timelines that would enable demonstrating the full and longer term benefits of the intervention. This is particularly important in RDs with nascent and borrowed endpoints that may fail to capture the full treatment benefit to patients and their families. Furthermore, despite the severe nature of many RDs and the fact that affected patients are often children, the disease impact or treatment benefit on caregivers and family is not routinely considered in HTAs [44].




Comments (0)