Study population
The RAPID-CPU registry is a monocenter observational study that enrolled consecutive patients presenting with suspected NSTE-ACS between July 1st, 2016, and June 30th, 2018, at the ED of Heidelberg University Hospital. A flow diagram for included and excluded patients within this study is shown in supplement Fig. 1. Details on the study population and interventions have been previously published [10]. Briefly, patients were eligible for enrollment if they presented with clinically suspected acute coronary syndrome (ACS), based clinically on a broad spectrum of symptoms including atypical chest pain or dyspnea. Exclusion criteria included missing 0-h or consecutive measurements if serial measurements were required, documented AV nodal re-entry tachycardia (AVNRT), acute heart failure due to known structural heart disease, primary pulmonary disease without suspected ACS, traumatic chest pain with preceding thorax injury, dysfunction or alarm of an implantable cardiac device (ICD), chronic hemodialysis, inadequate command of English/German language, or permanent residence in a foreign country. Patients were not excluded for severe chronic kidney disease, older age, chronic heart failure, suspected acute heart failure due to myocardial infarction (MI), atrial fibrillation, or missing 3-h measurements of the high-sensitivity cardiac troponin T (hs-cTnT) assay, as a third hs-cTn measurement at 3 h after the initial measurement was not obligatory until the 2015 ESC Guidelines [11].
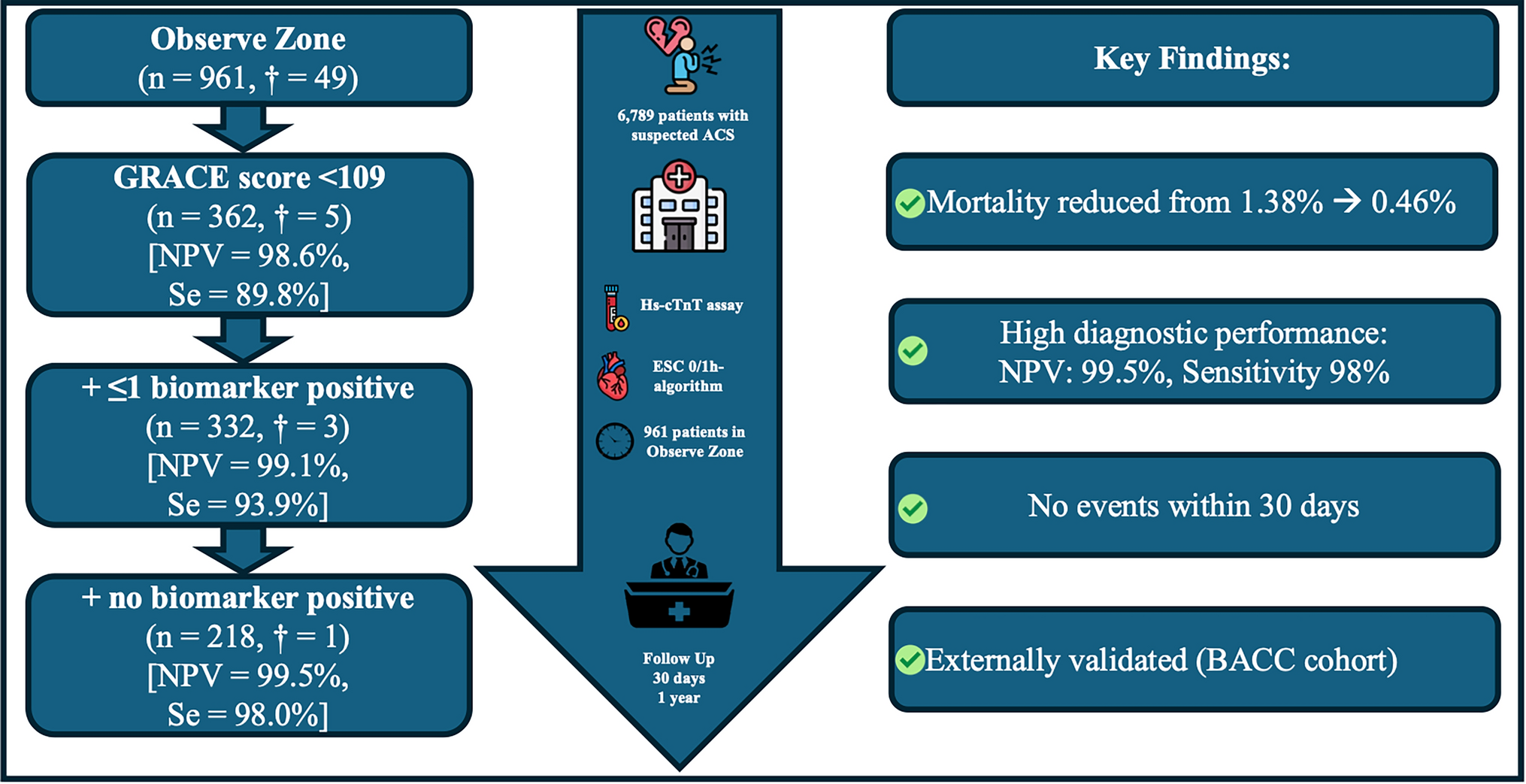
Using the validated ESC 0/1-h protocol, myocardial infarction was ruled out in patients presenting more than 3 h after symptom onset with an initial hs-cTnT below the limit of detection (LoD: < 5 ng/L), or if the initial hs-cTnT was < 12 ng/L with an absolute concentration change < 3 ng/L within the first hour. Patients were classified as 'rule-in' if the initial hs-cTnT concentration was ≥ 52 ng/L or if there was an absolute concentration change ≥ 5 ng/L within the first hour. Patients who did not fulfill either Rule-out or Rule-in criteria were categorized into the observe zone, and only these patients qualified for the present study. Diagnosis of MI was diagnosed by the treating physician using hs-cTnT and at that time of study conduct the criteria of the 2015 ESC guidelines [11], and the 4th version of the Universal Definition of Myocardial Infarction (UDMI) [12]. The GRACE 1.0 risk score was calculated using the Fox model for death between hospital admission and 6 months [9]. It integrates eight clinical parameters: age, heart rate, systolic blood pressure, serum creatinine, Killip classification, cardiac arrest at admission, ST-segment deviation on ECG, and elevated cardiac enzymes, following the original GRACE definitions. Scores of < 109, 109–140, and > 140 points categorize patients into low-, intermediate-, and high-risk groups, respectively.
External validation of the protocol was executed in the Biomarkers in Acute Cardiac Care (BACC) cohort which has been described earlier (Clinical Trials Identifier: NCT02355457) [13]. This observational study is an ongoing, prospective cohort study including patients who presented to the emergency department at the University Hospital of Hamburg with suspected non-ST-elevation acute coronary syndrome (NSTE-ACS).
Laboratory analyses
Plasma high-sensitivity cardiac troponin T (hs-cTnT) was measured with the Elecsys® Troponin T high-sensitivity assay (Roche Diagnostics) on a Cobas e411 immunoassay analyzer. LoB, LoD, 10% coefficient of variation (CV), and 99th percentile cut-off values were determined to be 3 ng/L, 5 ng/L, 13 ng/L and 14 ng/L [14, 15]. Copeptin in plasma samples at baseline (0 h) was measured with the copeptin proAVP assay on the KRYPTOR compact plus (BRAHMS Thermo Fisher Scientific). Detection limit, precision of 20% CV and 95th cut-off values for the copeptin proAVP assay were found to be 0.69 pmol/L, 1.08 pmol/L, and 9.8 pmol/L [16, 17]. An elevated copeptin was defined at concentrations > 10 pmol/L. NT-pro BNP was measured using the Siemens Atellica® using the general rule-out cutoff of 300 ng/L per ICON trial [24]. All other biomarkers including CRP (< 10 mg/dL), calculated eGFR (> 30 mL/min/1.73 m2), as well as hemoglobin (> 10 g/dL) were measured in the central laboratory on automated analyzers (Siemens) at established cutoffs [18,19,20,21]. The additional measurement of other biomarkers was not mandatory and was either part of laboratory routine, such as CRP, serum creatinine, eGFR and hemoglobin, or was ordered by the attending physician per clinical need for suspected comorbidities or underlying differential diagnoses of suspected ACS such as N-terminal pro-B-type natriuretic peptide (NT-proBNP) for suspected structural heart disease or acute heart failure, while D-dimer and copeptin were assessed for suspected venous thromboembolism, following the 2019 ESC guideline recommendations for pulmonary embolism [22] GFR was estimated based on serum creatinine using the race-independent CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) equation [23].
Follow-up
Patients were followed for a median of 12 months for the occurrence of all-cause death. Follow-up was accomplished using telephone, questionnaire, patient’s hospital notes, the family physician’s records, and the municipal registry on vital status. The study protocol was approved by the Ethics Committee of the Medical Faculty of Heidelberg. The trial was registered at ClinicalTrials.gov. (Clinical Trails.gov Identifier: NCT03111862).
Outcome and data collection
The primary outcome was 1-year all-cause mortality. This is generally considered the most useful outcome in identifying patients at very low risk of poor outcomes. Rates of cardiovascular (CV) death, MI, stroke, or other outcome events were not collected systematically. Data entry was performed by a dedicated research nurse, physician, or medical student at each site, and data collection included patient characteristics, clinical variables, and laboratory results at presentation required to calculate the GRACE-score. Data for determination of outcome measures were also collected.
Statistical analysis
Continuous variables were tested for normal distribution and were presented either as means with 95% CIs, or as medians with minimum and maximum. The normality of data distribution was assessed by the Kolmogorov–Smirnov test. Groups were compared using the χ2 test for categorical variables and Kruskal–Wallis test for continuous variables. Kaplan–Meier curves and the log-rank test were used. A classification and regression tree (CART) analysis was conducted with all-cause death as the primary outcome. The model identified GRACE 1.0 < 109 as the variable most strongly associated with outcomes. The absence of a positive biomarker or the presence of ≤ 1 biomarker served as the splitting points to best classify observations into groups. This combination of factors results in an easily visualized tree-like plot with corresponding event rates. Prevalence of individuals and events is provided for each tree branch. Our CART analysis focused on the model's ability to identify patients at the lowest risk, emphasizing the rule-out part. The model was developed using the entire training set, and the resulting tree structure was validated internally using bootstrapping and externally in the BACC cohort. All hypothesis testing was two-tailed and p values < 0.05 were considered statistically significant. All statistical analyses were carried out using the R software (version 4.3.0, R Foundation for Statistical Computing, Vienna, Austria) and MedCalc 20.111 (MedCalc Software bvba, Ostend, Belgium).


Comments (0)