Materials
FeCl2·4H2O, anhydrous FeCl3, dextran, ammonia solution, dimethyl sulfoxide (DMSO), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), and quercetin were purchased from Sigma-Aldrich. S. aureus (ATCC 25923) and S. epidermidis (ATCC 12228) were obtained from the Iranian Biological Resource Center. The culture media, buffers, and enzymes were also purchased from Sigma-Aldrich.
Fe3O4@Dex-QT synthesis
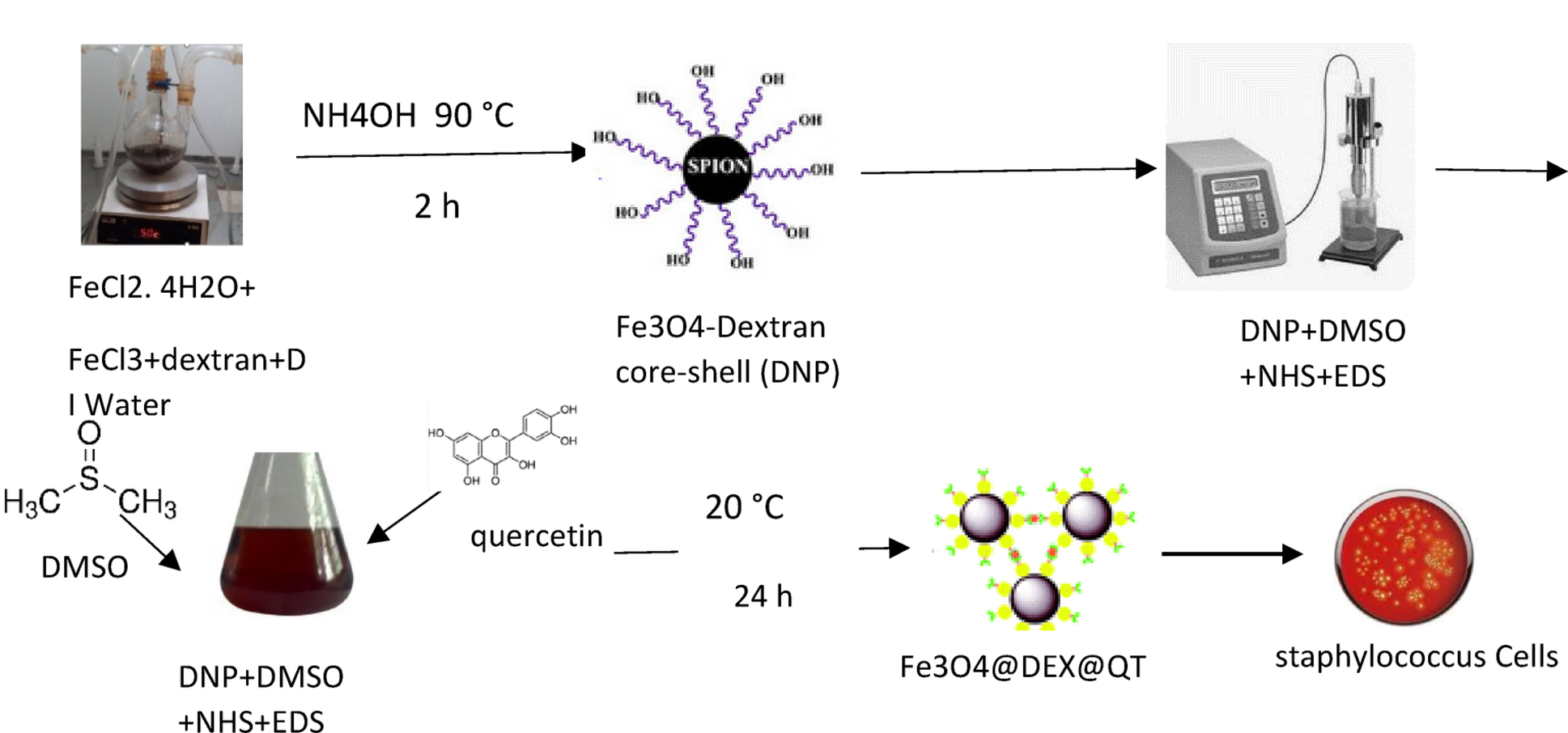
The synthesis of iron oxide nanoparticles was followed by a functionalization procedure of quercetin, as described by Yarjanli et al. (2019). A two-step method constitutes this process. The synthesis process of iron oxide nanoparticles commenced with dextran surface coating. A three-necked flask contained 1.135 g FeCl2.4H2O together with 0.695 g anhydrous FeCl3, and 0.45 g dextran while deionized water mixed with them through magnetic stirring. The mixture achieved pH level 9 when exposed to an ammonia solution, while performing continuous stirring at 90 °C for 2 h. A strong magnetic field extracted the resulting dextran-coated nanoparticles, before they received water and alcohol washing followed by thermal desiccation at 70 °C. Quercetin obtained success in binding to dextran-coated nanoparticles during their subsequent reaction phase. Ten milligrams of coated nanoparticles received dimethyl sulfoxide (DMSO) solution with N-hydroxysuccinimide (NHS), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) for required dosage. A mixture received sonication during 30 min before adding dissolving 10 mg of quercetin, into DMSO solution. The authors isolated the quercetin-functionalized nanoparticles by using powerful magnets and performed further analysis using FTIR, FE-SEM, and EDX.
Identification of S. aureus
Medical staff obtained blood samples in accordance with established clinical standards for staphylococcal infection analysis. The blood samples underwent aerobic and anaerobic culture bottle inoculation before researchers maintained them at 37 °C for five days. Microscopic analysis Gram-positive cocci that matched the morphology of Staphylococcus after applying Gram staining techniques. The research utilized ATCC 25923 as its standard clinical strain. The research employed tube coagulase test (TCT) and the thermonuclease test (TE test)-a agar diffusion method (ADM)- as two separate protocols to confirm Staphylococcus presence in blood isolates (Canning et al. 2020). A two-hour incubation period at 37 °C was applied to the 100 µL inoculated mixture, which contained broth mixed 0.5 mL of rabbit plasma under the TCT protocol. Clot formation during the process indicated that the test was positive. The negative tubes underwent an entire night of incubation, followed by a second examination the next day. Under the ADM approach, researchers performed boiling of 2 mL blood culture broth through for a 15-min period, followed by ambient temperature cooling. A solution of 60 µL liquid broth was added to six-millimeter well openings made in methyl green DNase agar plates. The laboratory incubated the plates at 37 °C for a duration of two hours. Researchers evaluated the agar diffusion method through observation of both dark green bands surrounding the wells and the clear zones within their vicinity. The positive control strain was S. aureus (ATCC: 25,923), along with the negative control strain being S. epidermidis (ATCC 12228) (Speers et al. 1998).
Minimum inhibitory concentration (MIC) assay
We used the broth microdilution test, as described in M07-A11 and adapted from Mokhtari et al. (2023) to determine the MIC for Fe₃O₄@Dex-QT nanoparticles. Quercetin-coated iron oxide nanoparticles were suspended in DMSO at 100 mg/mL and further diluted in CAMHB to prepare solutions with final concentrations ranging from 2 to 1024 μg/mL. Blood samples were cultured to obtain S. aureus strains, with one strain, ATCC 25923, used as a control. A bacterial suspension was prepared from overnight cultures on Mueller–Hinton agar and adjusted to 0.5 McFarland turbidity using an existing spectrophotometer at 625 nm. A 1:100 dilution of the suspensions was made in CAMHB to create a final inoculum of approximately 5 × 105 CFU/mL. One hundred microliters of the bacterial suspension were placed into each well containing varying concentrations of nanoparticles. The plates were incubated at 37 °C for 18 to 24 h. All assays were performed in duplicate to verify reproducibility. Bacterial growth was inhibited at the lowest concentration of nanoparticles, which marked the MIC.
Biofilm assay
Using the tissue culture plate approach outlined by Harika et al. (2020) and Mukherjee et al. (2017), this study conducted a biofilm assay to investigate both biofilm development and the inhibitory effects of Fe₃O₄@Dex-QT nanoparticles. Nineteen clinical S. aureus strains, along with the control strain ATCC 25923, were each grown in 5 mL TSB broth at 37 °C for 24 h. Fresh TSB was used to dilute cultures 1:100, ensuring the inoculum consistently contained approximately 1 × 10⁶ CFU/mL. Two hundred microliters of the diluted culture were pipetted into each well of a 96-well flat-bottomed plate (from Thermo Fisher Scientific). The concentrations determined from MIC testing (256 and 512 μg/mL) were used for Fe₃O₄@Dex-QT nanoparticles during anti-biofilm studies. To assess sterility and non-specific binding, wells without bacteria were used, and to demonstrate specific binding, wells with imipenem (at 10 μg/mL) were included and treated accordingly. The plates were sealed with adhesive lids and incubated at 37 °C for one day, ensuring air was present above the solutions to facilitate biofilm formation. During this stage, non-adherent cells were gently removed by aspirating the contents, and the wells were washed twice with 200 μL of PBS (pH 7.2) to remove non-adherent cells. A crystal violet solution was applied to each biofilm, and allowed to stain for 10 min. Up to two washing steps with deionized water removed excess stain, and the plates were dried at ambient temperature. The bound crystal violet was dissolved in 200 μL of 33% glacial acetic acid, and the optical density was measured at 630 nm (Bui et al. 2015) using a Bio-Rad 680 microplate reader. The choice of the 630 nm wavelength was based on the specifications of the equipment and confirmed for reliable performance in preliminary experiments. Assays were performed in triplicate, and the OD₆₃₀ was used to assess differences in biofilm formation and inhibition using one-way ANOVA. Both positive and negative controls were included to verify the assay’s specificity and efficiency.
Genetic analysis
We evaluated Coa and Hla gene expression levels through reverse transcription polymerase chain reaction (RT-PCR) after subjecting bacteria to the synthesized compounds.
RNA extraction
Total RNA was extracted from S. aureus using the RNA Extraction Kit (SinaClon, Iran, Lot No: 201903) following the manufacturer’s instructions, with modifications optimized for Gram-positive bacteria. Briefly, S. aureus cultures containing 2 × 10⁶ cells were centrifuged at 4,500 rpm for 10 min to pellet the cells. The supernatant was discarded, and the pellet was resuspended in 100 µL of G + Prelysis Buffer to facilitate cell wall disruption. Subsequently, 20 µL of lysozyme solution (15 mg/mL) was added, and the mixture was incubated at 37 °C for 20 min to enhance lysis of the Gram-positive cell wall. To remove contaminating genomic DNA, 10 µL of DNase I solution (1 U/µL) was added, and the samples were incubated at 55 °C for 30 min. Next, 400 µL of lysis buffer was added, and the samples were vortexed for 20 s. After adding 300 µL of binding solution and vortexing for 5 s, the mixture was centrifuged at 12,000 rpm for 1 min to bind RNA to the spin column. The column was washed with 400 µL of Wash Buffer II, followed by centrifugation at 12,000 rpm for 1 min. The column was then transferred to a clean microtube, and RNA was eluted in 30 µL of elution solution at 65 °C for 5 min, followed by centrifugation at 12,000 rpm for 1 min. The eluted RNA was stored at − 20 °C for short-term storage until further analysis. RNA concentration and purity were assessed using a Nanodrop spectrophotometer (Thermo Scientific), with A260/A280 ratios between 1.8 and 2.0 indicating high purity. RNA quality was further confirmed by successful amplification in downstream RT-qPCR experiments targeting Coa and Hla genes.
DNase treatment is used for cleaning RNA
To remove genomic DNA that might interfere, 1000 ng of RNA was treated with 1 µL of DNase I and 1 µL of 10X DNase I Buffer in a total volume of 20 µL, with the volume adjusted using nuclease-free water. All procedures were performed in accordance with the manufacturer’s guidelines, and after incubation, the RNA was purified.
cDNA synthesis
A cDNA library was created using the Synaclone kit. A 10 µL stock solution was prepared by mixing 7 µL of DEPC-treated water, 2 µL of RNA, and 1 µL of hexamer primer, then heating at 65 °C for 5 min and cooling on ice for at least 2 min. A second mixture was prepared, consisting of 2 µL of 10X M-MuLV buffer, 0.5 µL of M-MuLV reverse transcriptase, 0.5 µL of RNase inhibitor, 2 µL of 10 mM dNTPs, and 5 µL of DEPC-treated water. Combining the 10 µL mixture with the stock solution brought the total reaction volume to 20 µL. The Bio-Rad MyCycler thermocycler was used at 42 °C for one hour and 85 °C for five minutes to facilitate reverse transcription and produce cDNA from RNA.
Real time RT-PCR
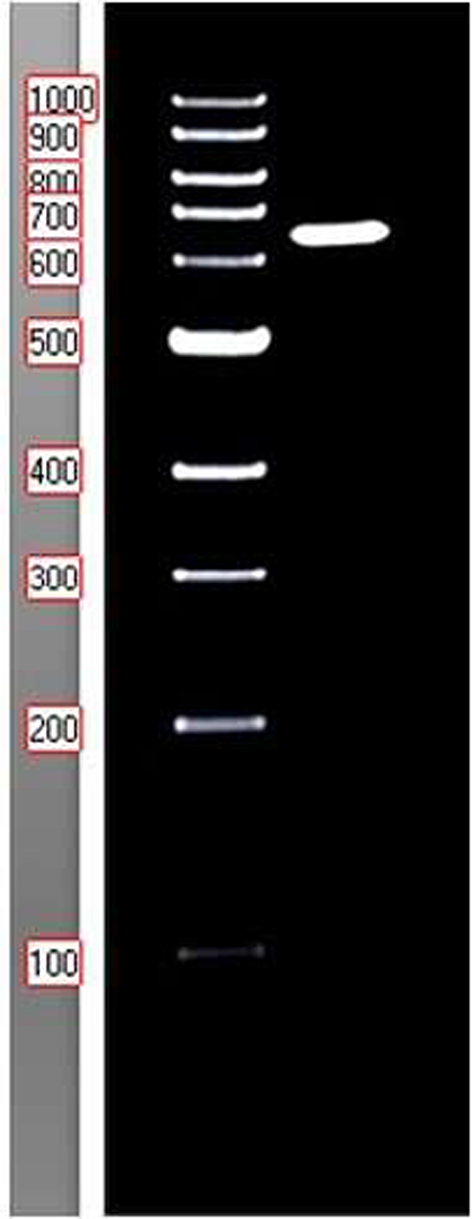
Real-time RT-PCR was performed to examine the expression levels of the Coa and Hla genes using primers from Javid et al. (2018) and Tavares et al. (2014). A 25 µL reaction mixture contained 0.5 µL of forward primer, 0.5 µL of reverse primer, 5 µL of SYBR Green, 1 µL of cDNA, and 3 µL of distilled water. The sample was subjected to the following cycling conditions: 95 °C for 30 s (initial hold), followed by 40 cycles of 95 °C for 20 s, 61 °C for 30 s, and 72 °C for 30 s, concluding with a melting curve from 60 °C to 95 °C. Three experiments were conducted for each analysis, and the corresponding dissociation curves confirmed the specificity of DNA strand amplification. Relative gene expression was calculated using the 2(ΔΔCt) method with 16S rRNA as the reference gene.
Coa F: ATAGAGATGCTGGTACAGG; R: GCTTCCGATTGTTCGATGC (Javid et al. 2018).
Hla F:CGAAAGGTACCATTGCTGGT: R: CCAATCGATTTTATATCTTTC (Tavares et al. 2014).
Statistical analysis
ANOVA tests with SPSS software version 23 analyzed variances (p < 0.05) between treated and untreated groups.


Comments (0)