
Remember me

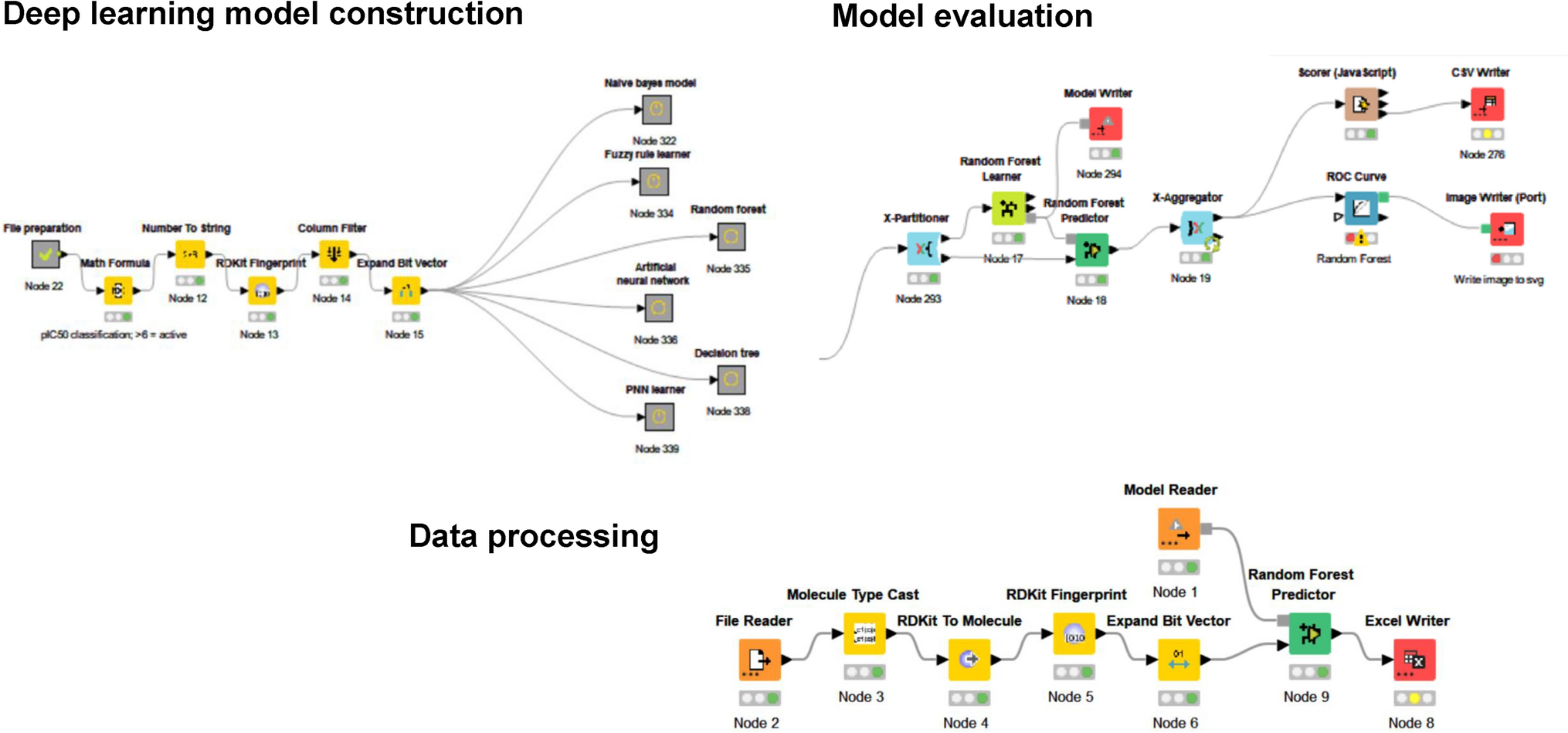

Comparison of the experimental structures shows that there is little difference between the ligand-bound structures and the ligand-free structure, suggesting that HDAC6 is a rather rigid structure (see Table I).
Table I RMSDs between ligand-free structure (5EEM, chain A) and ligand-bound structures after least-squares superposition of Cα atomsPCA on ligand-free equilibrium trajectory of HDAC6The 200 ns trajectory of the HDAC6 receptor was represented by 2000 frames. An RMSD plot of the conformations from frames 1-2000 fitted to frame 1 showed that during the first 20 ns, the protein was still equilibrating (see Fig. S1). We therefore performed PCA on frames 201–2000. Note that there is a more gradual increase in the RMSD from about 120 ns to the end of the simulation at 200 ns. This is possibly due to regions of the protein, such as loops, exhibiting slow relaxation. PCA was performed on all protein atoms and the zinc ion, but water molecules and ions, including the two crystallographic potassium ions, were not included. This amounted to 5478 atoms. This all-atom PCA was used for the linear response as described in previous publications [20, 28, 29] and applied to the HDAC6 trajectory. Figure 4 (A) shows the trajectory projected onto the first two principal modes which shows three distinct clusters. Equivalent figures by Shahab et al. [17] for 100 ns trajectories of HDAC6 complexes also indicated the presence of clusters although more than the three seen here. The first two principal modes (out of a total 16428) contribute 16% and 6%, respectively, to the total MSF. As described in a recent review [39] the conformations were projected onto the first and second modes (PC’s) to find the maximum and minimum extent of the modes. This produced two structures for each mode, \(\:}_^\) and \(\:}_^\) for mode 1, and \(\:}_^\) and \(\:}_^\) for mode 2. Although HDAC6 does not visually seem to comprise structural domains, these maximum and minimum structures were input into the DynDom program [41] at the DynDom webserver [42], which revealed a clear domain movement for mode 1 (PC1), but not for mode 2 (PC2). The resulting dynamic domains, hinge axis, and hinge bending regions are shown in Fig. 5. The domain boundary bisects the binding pocket and although the angle of rotation about the hinge axis is small (8.2°), the movement produces an appreciable widening and narrowing of the interdomain cleft at the active site as can be seen in Fig. 5. The movement in the positive direction of mode 1 causes the binding pocket to widen and in the negative direction it causes the pocket to narrow (see Fig. 4 (A)). Interdomain bending residues are: 444–448, 482–484, 551–552, 564–565, 615–618, 739–740, and 768–772 (note DockIT residue numbering is PDB residue numbering minus 441). The movement is 77% a twisting motion [43] controlled by the central β-sheet with the hinge axis on the surface of the sheet, oriented perpendicular to the strands, and passing close to the interdomain bending region on each strand. Like many proteins that undergo a small twisting of their domains, the movement is classified as predominantly shear [44] indicating that there is a relative sliding of the domains at their interface.
Fig. 4
The black points are the 1800 frames from 20.1 ns-200 ns portion of trajectory projected on to the first two principal modes, PC1 and PC2. The red line shows a typical docking trajectory. Automated docking is indicated by a continuous line, and manual docking by a broken line. The blue filled square is the relaxed receptor conformation. The open blue squares correspond to the different starting poses. Purple circles are at the intermediate binding site where the side chain hydroxyl group of Tyr745 forms a hydrogen bond with the hydroxamic acid group. Open red squares are immediately after the push over energy barrier and green squares (all coinciding) indicate the final docked conformation (A) Movement in the direction of negative values for PC1 cause the binding pocket to narrow. Conversely movement in the direction of positive values for PC1 causes the binding pocket to widen (B) Docking of belinostat for the 14 cases (C) Docking of HPOB: the 7 cases where the hydroxamic acid group is oriented as in the HPOB-bound crystallographic structure (D) Docking of HPOB: the 5 cases where the hydroxamic acid group is in the same orientation as in belinostat
Fig. 5
DynDom result where input is the two extreme projections \(\:}_^\) and \(\:}_^\) on the first principal mode from the PCA of the HDAC6 MD trajectory. The dynamic domains are in red and blue, hinge bending regions in green. In stick depiction is the belinostat ligand fitted into the active site. The domain boundary bisects the binding pocket indicating that the domain motion affects its size and shape. (A) Cartoon depiction showing hinge axis as an arrow (B) At the minimum value, \(\:}_^\) (C) At the maximum value structure, \(\:}_^\), where the binding pocket has widened in comparison to the \(\:}_^\) structure
As already stated, to evaluate the response in real time we use the first M principal modes. Here we use M = 100, which accounted for 66% of the total MSF. Regarding the stability of this subspace, we divided the trajectory on which the PCA was performed into two equal halves and performed PCA on each. The root mean-square inner product (RMSIP) is a measure of the overlap of two subspaces and for identical subspaces (100% overlapping) achieves its maximum value of 1.0. For M = 100, the RMSIP value was 0.62.
In addition to the all-atom PCA used for linear response, we also performed a PCA on just backbone atoms (N, Cα and C) so that we could easily compare experimental structures in the subspace of the first two principal modes. In Supplementary Material Fig. S2 we present the crystallographic ligand-free, belinostat and HPOB bound structures projected on the first two principal modes of the backbone PCA. It shows that both the structures are closely located within the two clusters on the left in Fig. 4 (A) (which are somewhat merged in the backbone PCA). Thus, both experimental structures and the ligand-free structure are associated with the clusters on the left which have a narrower binding pocket than the cluster on the right.
The first and second principal modes (out of a total 3198) for this backbone PCA contribute 34% and 7% to the total MSF, respectively, indicating a very dominant first principal mode. A DynDom analysis for the movement between the minimum and maximum structures along mode 1 produced an almost identical result to the all-atom case. The RMSIP for M = 100 over the two halves of the backbone atom trajectory is 0.84, indicating a rather stable subspace.
To address the possible issue of whether a single MD simulation of 200 ns provides sufficient sampling, we performed two additional 200 ns simulations and evaluated the backbone RMSIPs between their 100-dimensional subspaces. The RMSIP values were 0.81 and 0.77, indicating a relatively stable subspace. This is in accordance with a previous study [29] where the docking-induced domain movement in maltodextrin binding protein and glutamine binding protein (MBP is the same size as HDAC6, GBP is slightly smaller) showed an excellent agreement with its respective experimentally determined domain movement even though the subspace for the linear response was derived from a shorter 100 ns trajectory.
Initial placement of inhibitor molecules for automated dockingFor both inhibitors, the following procedure was carried out to find positions from which to start automated docking. The molecule was inserted into the binding pocket and then moved so that the hydroxamic acid was clearly outside of the pocket. If it moved back into the binding pocket when automated docking was engaged, then this position was used as a starting position from which a new position and orientation was trialed by implementing a small translation and rotation of the drug molecule. If from this new position the drug entered the binding pocket, this became the new starting position. This process was repeated 40–50 times for each molecule. Belinostat entered the binding cavity in 14 out of 46 trial positions, and HPOB entered the binding cavity in 12 out of 43 trials. Figure 6 shows the starting positions of both inhibitors for those starting positions that entered the cavity.
Fig. 6
Shows the starting poses of inhibitors (stick depiction) that successfully entered the binding pocket of HDAC6 (molecular surface depiction). The red arrow indicates the binding pocket. (A) The 14 starting poses for belinostat (B) The 12 starting poses for HPOB. The 7 in cyan form a hydrogen bond to Tyr745, the hydroxamic acid group oriented as found in the crystallographic structure (PDB:5EF7). The 5 in magenta rotate almost 180° about the long axis of the hydroxamic acid group and form hydrogen bonds with Tyr745 and Asn530
Docking of belinostatThe 14 starting positions, shown in Fig. 6 (A), from which belinostat entered the binding pocket, had RMSDs with the ghost at the experimentally bound position in the range 8.7–16.2 Å. In Fig. 4 (B) the conformation of HDAC6 is projected onto the plane of the first two principal modes for selected positions of belinostat relative to HDAC6 during binding. It shows that HDAC6 changes conformation during the binding process. In all 14 cases, the final position of belinostat after automated docking resulted in the same binding pose with belinostat hydrogen bonded to the side chain hydroxyl group of Tyr745 as shown in Fig. 7 (A). During this stage Fig. 4 (B) indicates that domain twisting acts to narrow the binding pocket. Visually comparing this conformation with the relaxed conformation confirmed that a narrowing of the pocket had occurred and a DynDom analysis of the movement from the relaxed receptor conformation to this Tyr745 bound conformation showed an almost identical domain decomposition to that seen in Fig. 5, with the hinge axis pointing approximately in the opposite direction indicating narrowing of the binding pocket for this domain-twisting movement. For those starting positions further away from HDAC6, belinostat first forms a hydrogen bond with the side chain of Asn530, then forms a hydrogen bond with Asn645, before finally hydrogen bonding with Tyr745. Comparing the Tyr745 hydrogen bonded pose of belinostat with its ghost in the binding pocket reveals that it is at an intermediate binding site where it is partially inserted into the binding pocket (3.6 Å RMSD with the ghost). As this is at an energy minimum (total energy is -28 kcal/mol), there must be an energy barrier between the Tyr745 hydrogen-bonded pose and the crystallographic binding pose. Switching back to manual mode, belinostat was gently pushed further into the binding pocket causing an increase in the strain energy. In doing this, it forms a hydrogen bond with the main chain of Phe643, and the Leu712 moves away from the pocket mainly through the movement of the loop 709–716, on the tip of which Leu712 is located. After this manual intervention, where the RMSD with the ghost is 2.7 Å, another round of automated docking was implemented in which belinostat breaks its hydrogen bond with Phe643 and moves to its final binding pose that has a 1.0 Å RMSD with the ghost, i.e., very close to the experimentally determined binding pose (total energy is -40 kcal/mol), (see Fig. 7 (B)). During this stage the interaction and strain energies decrease. This relaxation results in a domain twisting that acts this time to widen the binding pocket (see Fig. 4 (B)). The estimated height of the energy barrier between the intermediate pose and the final binding pose is ∼ 10 kcal/mol.
Fig. 7
In purple stick depiction is the belinostat ligand. Green stick depiction is Tyr745. Hydrogen bonds are indicated by broken yellow lines and the zinc ion is depicted as a grey sphere (A) Belinostat in the intermediate pose after first phase of automated docking showing its hydrogen bonds with Tyr745 (B) Belinostat ligand has moved closer to the zinc ion in its final binding pose which is close to its crystallographic binding pose depicted in cyan stick
It is to be noted that this final binding pose is identical to when belinostat is superimposed on its ghost and automated docking engaged until an energy minimum is found.
There are some observations that can be made about this process. For some paths to the intermediate binding site, there are simultaneous hydrogen bonds between Asn645 and the sulfonamide group of belinostat and between Tyr745 and the hydroxamic acid group of belinostat suggesting a relay of interactions occurs during binding. From the path shown in Fig. 4 (B) one can see that as belinostat moves into the binding pocket it is accompanied by domain twisting along the first principal mode of HDAC6 that acts to narrow the size of the pocket. Interestingly, the conformation of HDAC6 moves outside the distribution of conformations seen for the ligand-free protein, indicating an induced-fit mechanism. In these conformations the HDAC6 closes further upon belinostat with the binding pocket adjusting size so that it snuggly fits to belinostat as judged by their complementary molecular surfaces. The docking trajectories for the conformation of HDAC6 do not approach conformations in the cluster on the right in Fig. 4 (B), and like the experimental structures (see Fig. S2), the final docked structure of HDAC6 is very close to the relaxed, ligand-free receptor conformation (RMSD = 0.09 Å, calculated on Cα atoms for comparison to the experimental values given in Table I).
Docking of HPOBThe 12 starting positions, shown in Fig. 6 (B), from which HPOB entered the binding pocket, had RMSDs with the HPOB ghost at the crystallographic bound position in the range 9.7–14.2 Å. In Fig. 4 (C) and (D) the conformation of HDAC6 during the binding process is projected onto the plane of the first two principal modes for selected poses of HPOB relative to HDAC6. As for belinostat, after automated docking, HPOB moves to an intermediate binding pose at an energy minimum where a hydrogen bond with the side chain hydroxyl group of Tyr745 results. The movement to this intermediate site is, as with belinostat, also accompanied by a domain twisting that acts to narrow the size of the binding pocket. Interestingly the 12 intermediate binding poses can be divided into two distinct groups (see Fig. 6 (B)), 7 with the hydroxamic acid group oriented as found in the crystallographic HPOB-HDAC6 complex structure (total energy is -25 kcal/mol) and 5 with the hydroxamic acid group, oriented in as in belinostat (total energy is -30 kcal/mol). HPOB in the former group will be referred to as being in the “crystallographic orientation” and the latter group will be referred to as being in the “belinostat orientation.” In this intermediate binding pose the hydroxamic acid in the crystallographic orientation is an almost exact 180° flip of its orientation in the belinostat orientation.
HPOB that binds in the crystallographic orientationAt the intermediate pose with the hydrogen bond between HPOB and Tyr745, HPOB had a 2.4 Å RMSD with the HPOB ghost (see Fig. 8 (A)). As for belinostat we gave HPOB a gentle push further into the pocket from this intermediate pose. After this manual intervention the RMSD of HPOB with ghost HPOB was 1.6 Å. After re-engaging automated docking, they all moved to deeper into the pocket to reach a final binding pose that had a 1.4 Å RMSD with the ghost, i.e., very close to the crystallographic binding pose (the total energy is -35 kcal/mol) (see Fig. 8 (B)). As for belinostat, during this stage domain twisting acts to widen the binding pocket (see Fig. 4 (C)). The estimated height of the energy barrier between the intermediate pose and the final binding pose is ∼ 2–3 kcal/mol.
Fig. 8
In purple stick depiction is the HPOB ligand. Green stick depiction is Tyr745. Hydrogen bonds are indicated by broken yellow lines and the zinc ion is depicted as a grey sphere (A) HPOB with its hydroxamic acid in the crystallographic orientation in the intermediate site having formed a hydrogen bond with Tyr745 (B) Final docked binding pose of HPOB with hydroxamic acid in the crystallographic orientation compared with the crystallographic pose of HPOB (cyan stick) (C) HPOB in with its hydroxamic acid in the same orientation as belinostat in the intermediate site with a hydrogen bond to Tyr745. It also formed a hydrogen bond with Asn530 (not shown). Note the orientation of HPOB in (A) and (B) is approximately a 180° flip about the long axis of the hydroxamic acid group in comparison to its orientation in (C)
This final binding pose is identical to when HPOB is superimposed on its ghost and automated docking engaged until an energy minimum is found.
HPOB that binds in belinostat orientationAt the intermediate pose, HPOB in the belinostat orientation had a 5.0 Å RMSD with ghost HPOB (Fig. 8 (C)). In addition to the hydrogen bond with Tyr745 it had a hydrogen bond with the side chain of Asn530. Pushing HPOB further into the pocket and re-engaging automated docking, resulted in it moving back out of the pocket, indicating a very high energy barrier. The likely reason for HPOB not being able to fully enter the binding pocket in this orientation is that steric clashes between the cap group and the aperture of the HDAC6 binding pocket prevent it.
Role of cap group of HPOB in determining orientationIf we define a “contact” to be when any pair of heavy atoms are within 4 Å of each other, and the cap of HPOB to comprise atoms O3, O4, N2, and C6-C15 (atom names from PDB file: 5EF7), then with HPOB at the intermediate binding site, there are 9 contacts between the cap of HPOB and HDAC6 in the belinostat orientation, compared to only 4 for the crystallographic orientation. In the crystallographic orientation there is space between the cap and HDAC6 allowing HPOB to move deeper into the binding pocket (see Fig. 9 (A)). The contacts for HPOB in the belinostat orientation that appear to block further penetration deeper into the pocket are primarily between the O-N-C2-OH group of HPOB and Asn530 of HDAC6 (see Fig. 9 (B)). From these results it is tempting to draw the conclusion that HPOB binds with its hydroxamic acid in a flipped orientation compared to belinostat because there is a significantly lower energy barrier between the intermediate pose and the fully inserted pose due to there being fewer intermolecular contacts between the HPOB cap and HDAC6 in this orientation.
Fig. 9
Molecular surface depictions of HDAC6 (green) and HPOB (purple) at the intermediate binding site. (A) In crystallographic orientation from which it is able to move deeper into the pocket (B) In belinostat orientation where it is unable to move deeper into the pocket due to the greater number of contacts between the cap and HDAC6
Comments (0)