Van Drie JH (2007) Computer-aided drug design: the next 20 years. J Comput Aided Mol Des 21(10–11):591–601
CAS
PubMed
Google Scholar
Stanzione F, Giangreco I, Cole JC (2021) Use of molecular docking computational tools in drug discovery. Prog Med Chem 60:273–343
PubMed
Google Scholar
Koukos PI, Xue LC, Bonvin AM (2019) Protein-ligand pose and affinity prediction: lessons from d3r grand challenge 3. J Comput Aided Mol Des 33:83–91
CAS
PubMed
Google Scholar
Yang C, Chen EA, Zhang Y (2022) Protein-ligand docking in the machine-learning era. Molecules 27(14):4568
CAS
PubMed
PubMed Central
Google Scholar
Hassan NM, Alhossary AA, Mu Y, Kwoh C-K (2017) Protein-ligand blind docking using quickvina-w with inter-process spatio-temporal integration. Sci Rep 7(1):15451
PubMed
PubMed Central
Google Scholar
Sahu MK, Nayak AK, Hailemeskel B, Eyupoglu OE (2024) Exploring recent updates on molecular docking: types, method, application, limitation & future prospects. Int J Pharm Res Allied Sci 13(2–2024):24–40
Google Scholar
Ugurlu SY, McDonald D, Lei H, Jones AM, Li S, Tong HY, Butler MS, He S (2024) Cobdock: an accurate and practical machine learning-based consensus blind docking method. J Cheminf 16(1):5
Google Scholar
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998) Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19(14):1639–1662
CAS
Google Scholar
Trott O, Olson AJ (2010) Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
CAS
PubMed
PubMed Central
Google Scholar
Vorobjev YN (2010) Blind docking method combining search of low-resolution binding sites with ligand pose refinement by molecular dynamics-based global optimization. J Comput Chem 31(5):1080–1092
CAS
PubMed
Google Scholar
Hetényi C, Spoel D (2002) Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci 11(7):1729–1737
PubMed
PubMed Central
Google Scholar
Hetényi C, Spoel D (2006) Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett 580(5):1447–1450
PubMed
Google Scholar
Huang Y, Zhang H, Jiang S, Yue D, Lin X, Zhang J, Gao YQ (2023) Dsdp: a blind docking strategy accelerated by gpus. J Chem Inf Model 63(14):4355–4363
CAS
PubMed
Google Scholar
Yan J, Zhang Z, Zhu J, Zhang K, Pei J, Liu Q (2024) Deltadock: a unified framework for accurate, efficient, and physically reliable molecular docking. arXiv preprint arXiv:2410.11224
Shen A, Yuan M, Ma Y, Du J, Wang M (2024) Pgbind: pocket-guided explicit attention learning for protein-ligand docking. Brief Bioinform 25(5):455
Google Scholar
Ghersi D, Sanchez R (2009) Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins 74(2):417–424
CAS
PubMed
PubMed Central
Google Scholar
Liu Y, Grimm M, Dai W-T, Hou M-C, Xiao Z-X, Cao Y (2020) Cb-dock: a web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol Sin 41(1):138–144
PubMed
Google Scholar
Wu Q, Peng Z, Zhang Y, Yang J (2018) Coach-d: improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res 46(W1):438–442
Google Scholar
Liu Y, Yang X, Gan J, Chen S, Xiao Z-X, Cao Y (2022) Cb-dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res 50(W1):159–164
Google Scholar
Krivák R, Hoksza D (2018) P2rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J Cheminf 10:1–12
Google Scholar
Le Guilloux V, Schmidtke P, Tuffery P (2009) Fpocket: an open source platform for ligand pocket detection. BMC Bioinf 10(1):1–11
Google Scholar
Corso G, Stärk, H, Jing B, Barzilay R, Jaakkola T (2022) Diffdock: diffusion steps, twists, and turns for molecular docking. arXiv preprint arXiv:2210.01776
Lu W, Zhang J, Huang W, Zhang Z, Jia X, Wang Z, Shi L, Li C, Wolynes PG, Zheng S (2024) Dynamicbind: predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model. Nat Commun 15(1):1071
CAS
PubMed
PubMed Central
Google Scholar
Du J, Yuan M, Shen A, Wang M (2025) Ppdock: pocket prediction-based protein-ligand blind docking. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.4c01373
PubMed
Google Scholar
Masters M, Mahmoud AH, Lill MA (2023) Pocketnet: ligand-guided pocket prediction for blind docking. In: ICLR 2023-Machine Learning for Drug Discovery Workshop
Zhang W, Bell EW, Yin M, Zhang Y (2020) Edock: blind protein-ligand docking by replica-exchange Monte Carlo simulation. J Cheminf 12:1–17
Google Scholar
Hernandez M, Ghersi D, Sanchez R (2009) Sitehound-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res 37(suppl–2):413–416
Google Scholar
Jiménez J, Doerr S, Martínez-Rosell G, Rose AS, De Fabritiis G (2017) Deepsite: protein-binding site predictor using 3d-convolutional neural networks. Bioinformatics 33(19):3036–3042
PubMed
Google Scholar
Zhang Z, Li Y, Lin B, Schroeder M, Huang B (2011) Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 27(15):2083–2088
CAS
PubMed
Google Scholar
Huang B (2009) Metapocket: a meta approach to improve protein ligand binding site prediction. OMICS 13(4):325–330
CAS
PubMed
Google Scholar
Fang Y, Jiang Y, Wei L, Ma Q, Ren Z, Yuan Q, Wei D-Q (2023) Deepprosite: structure-aware protein binding site prediction using esmfold and pretrained language model. Bioinformatics 39(12):718
Google Scholar
Tubiana J, Schneidman-Duhovny D, Wolfson HJ (2022) Scannet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nat Methods 19(6):730–739
CAS
PubMed
Google Scholar
Sun X, Wu Z, Su J, Li C (2024) Graphpbsp: protein binding site prediction based on graph attention network and pre-trained model prostt5. Int J Biol Macromol 282:136933
CAS
PubMed
Google Scholar
Seo S, Choi J, Choi S, Lee J, Park C, Park S (2024) Pseq2sites: enhancing protein sequence-based ligand binding-site prediction accuracy via the deep convolutional network and attention mechanism. Eng Appl Artif Intell 127:107257
Google Scholar
Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J et al (2024) Addendum: accurate structure prediction of biomolecular interactions with alphafold 3. Nature 636:1
Google Scholar
Halgren TA (2009) Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 49(2):377–389
CAS
PubMed
Google Scholar
Torres PH, Sodero AC, Jofily P, Silva-Jr FP (2019) Key topics in molecular docking for drug design. Int J Mol Sci 20(18):4574
CAS
PubMed
PubMed Central
Google Scholar
Jofily P, Pascutti PG, Torres PH (2021) Improving blind docking in dock6 through an automated preliminary fragment probing strategy. Molecules 26(5):1224
CAS
PubMed
PubMed Central
Google Scholar
Chen Y-C (2015) Beware of docking! Trends Pharmacol Sci 36(2):78–95
PubMed
Google Scholar
Mo Q, Xu Z, Yan H, Chen P, Lu Y (2023) Vsth: a user-friendly web server for structure-based virtual screening on tianhe-2. Bioinformatics 39(1):740
Google Scholar
Ugurlu SY, McDonald D, He S (2024) Mef-allosite: an accurate and robust multimodel ensemble feature selection for the allosteric site identification model. J Cheminf 16(1):116
CAS
Google Scholar
Zhao S, Zhang Y, Xu H, Han T (2019) Ensemble classification based on feature selection for environmental sound recognition. Math Probl Eng 2019(1):4318463
Google Scholar
Dong J, Yao Z-J, Zhang L, Luo F, Lin Q, Lu A-P, Chen AF, Cao D-S (2018) Pybiomed: a python library for various molecular representations of chemicals, proteins and dnas and their interactions. J Cheminf 10:1–11
Google Scholar
Chapman B, Chang J (2000) Biopython: python tools for computational biology. ACM Sigbio Newslett 20(2):15–19
Google Scholar
Bonidia RP, Domingues DS, Sanches DS, Carvalho AC (2022) Mathfeature: feature extraction package for dna, rna and protein sequences based on mathematical descriptors. Brief Bioinform 23(1):434
Google Scholar
Exner TE, Korb O, Ten Brink T (2009) New and improved features of the docking software plants. Chem Cent J 3(1):1–1
Google Scholar
Bredel M, Jacoby E (2004) Chemogenomics: an emerging strategy for rapid target and drug discovery. Nat Rev Genet 5(4):262–275
CAS
PubMed
Google Scholar
Zhang W, Huang J (2022) Evis: an enhanced virtual screening approach based on pocket-ligand similarity. J Chem Inf Model 62(3):498–510
CAS
PubMed


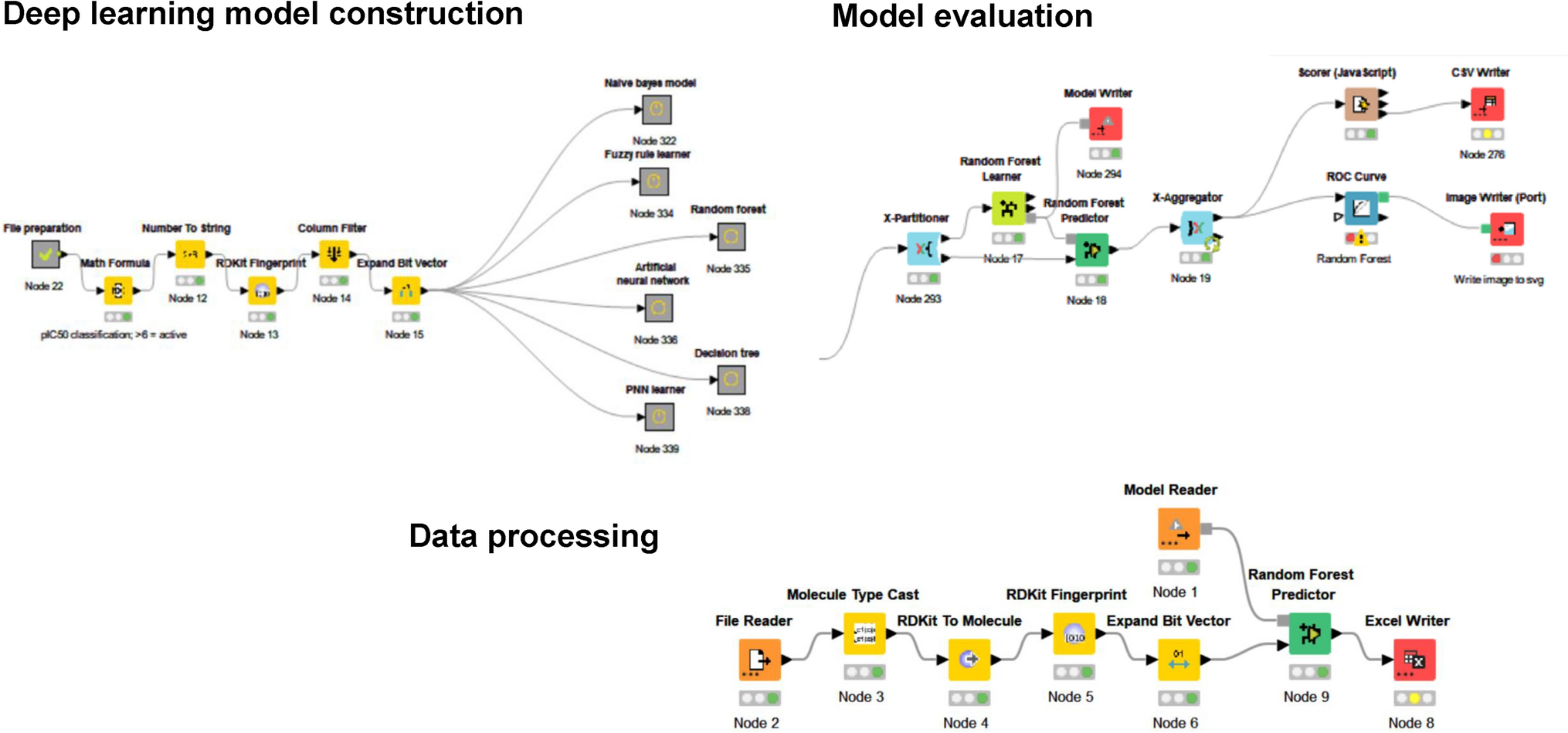
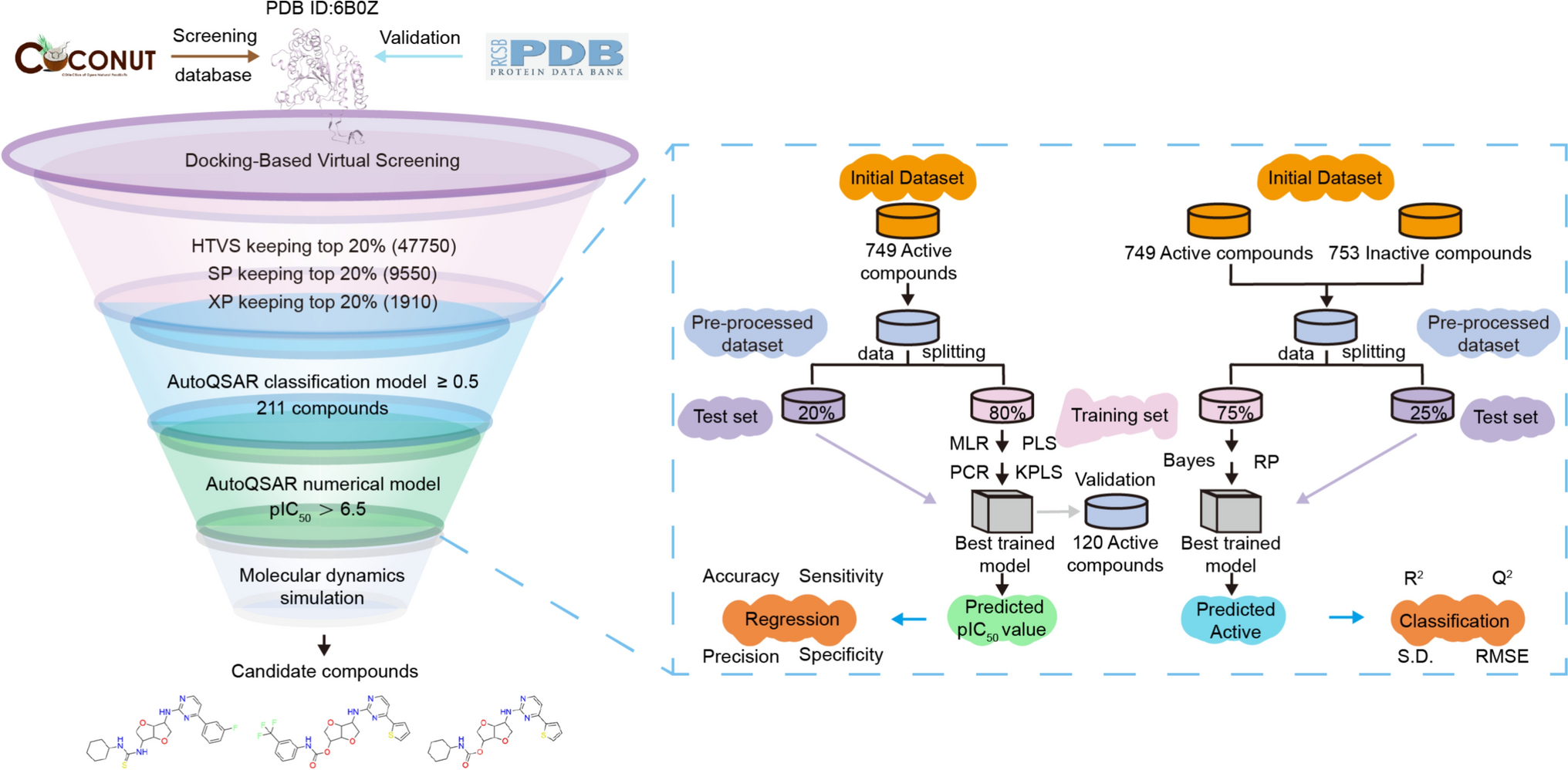

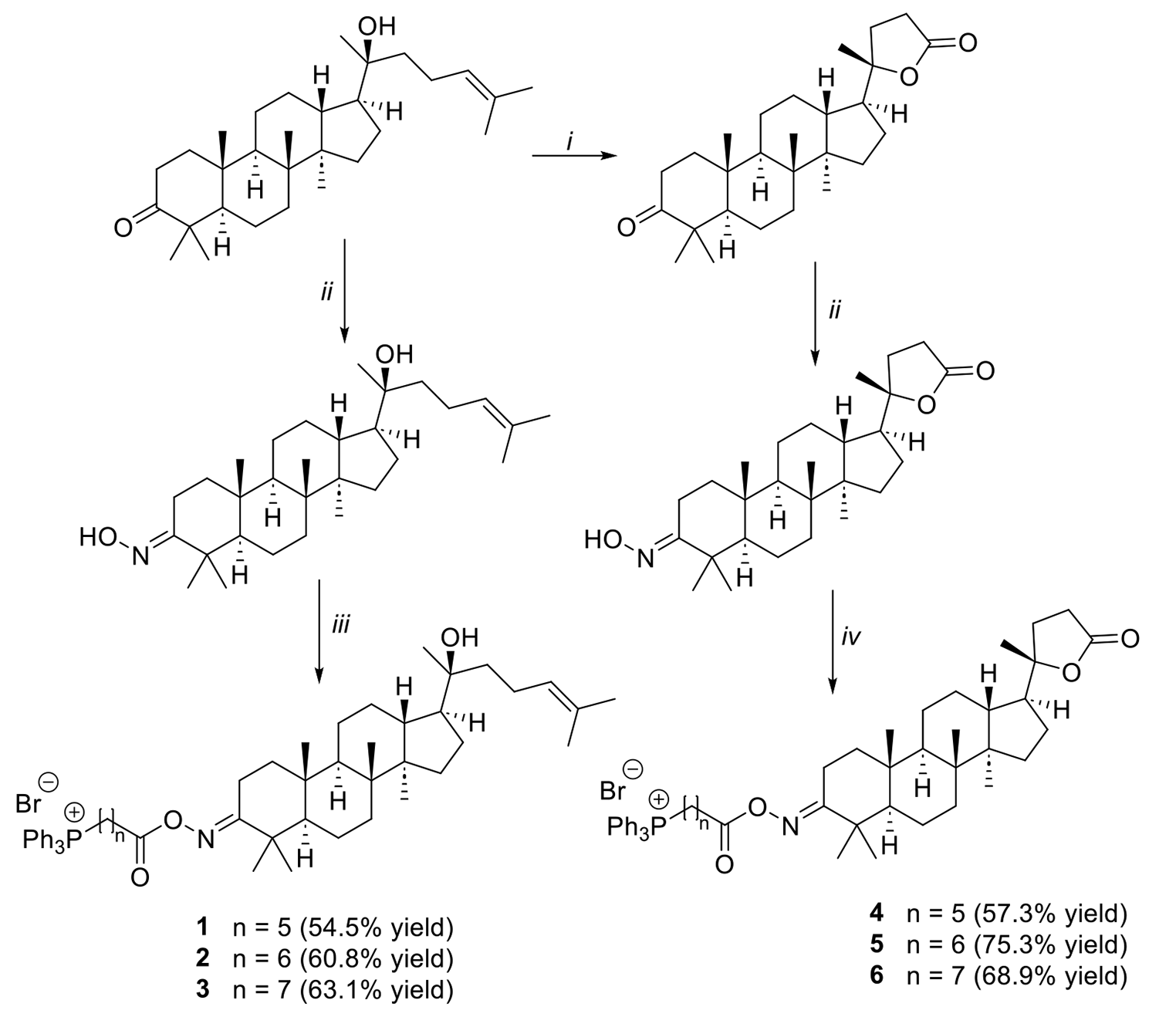
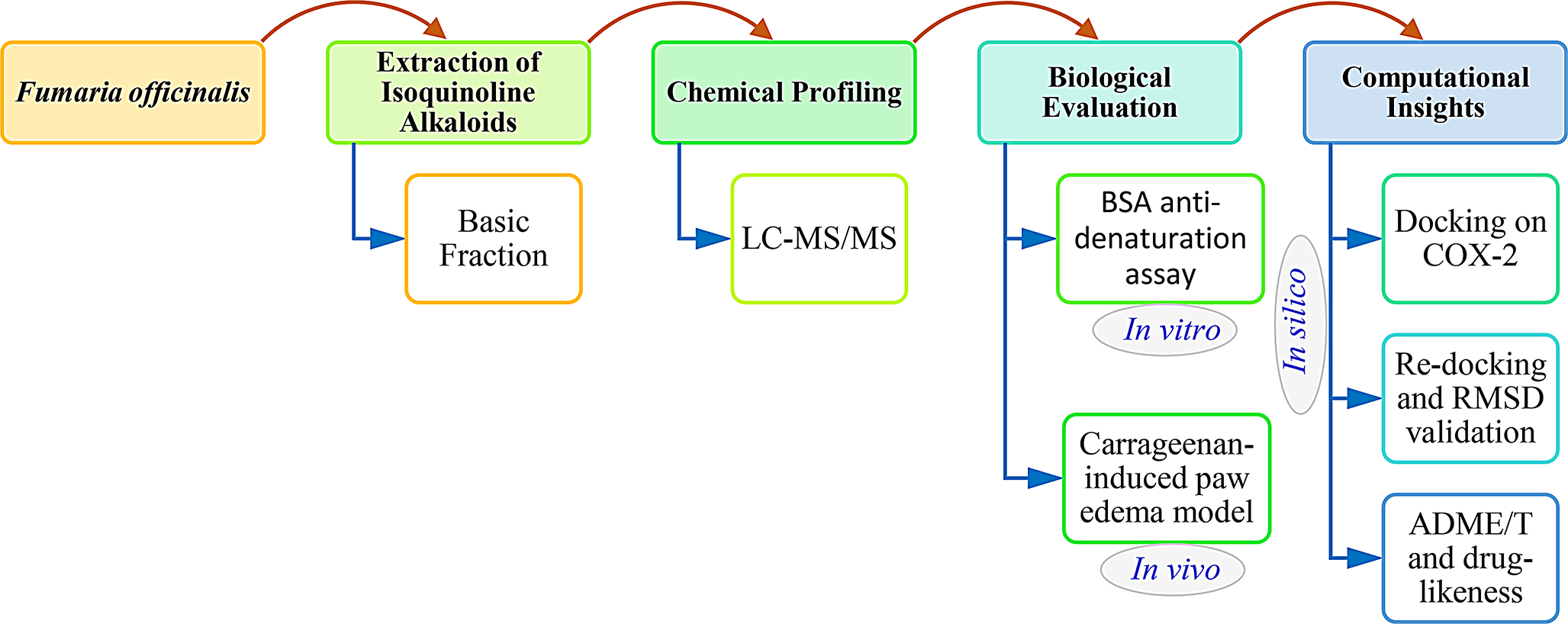
Comments (0)