
Remember me
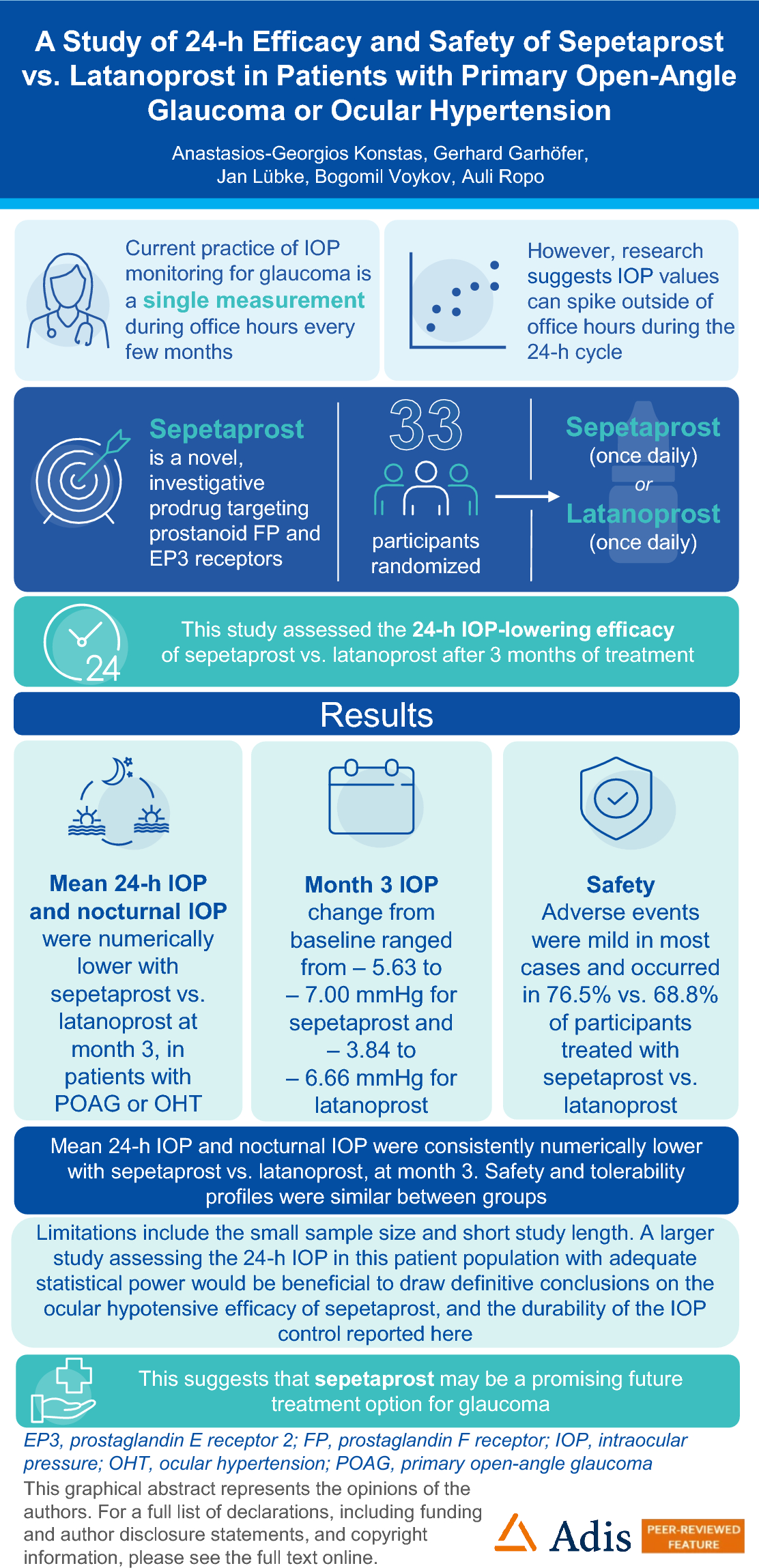



This interchangeability study (Clinicaltrial.gov identifier: NCT05637515, EudraCT No:2021–006015-29) enrolled eligible patients from 31 sites across 4 countries in Eastern Europe, Poland, Bulgaria, Estonia, and Czech Republic, between 5-December-2022 and 19-September-2023. It was conducted in accordance with the Declaration of Helsinki, in compliance with the International Council for Harmonization, Good Clinical Practice guidelines, and as per national, state and local regulatory authorities. All the study-related documents were reviewed by independent ethics committees and institutional review boards for each study center. All patients signed and dated the informed consent form before participating in the study.
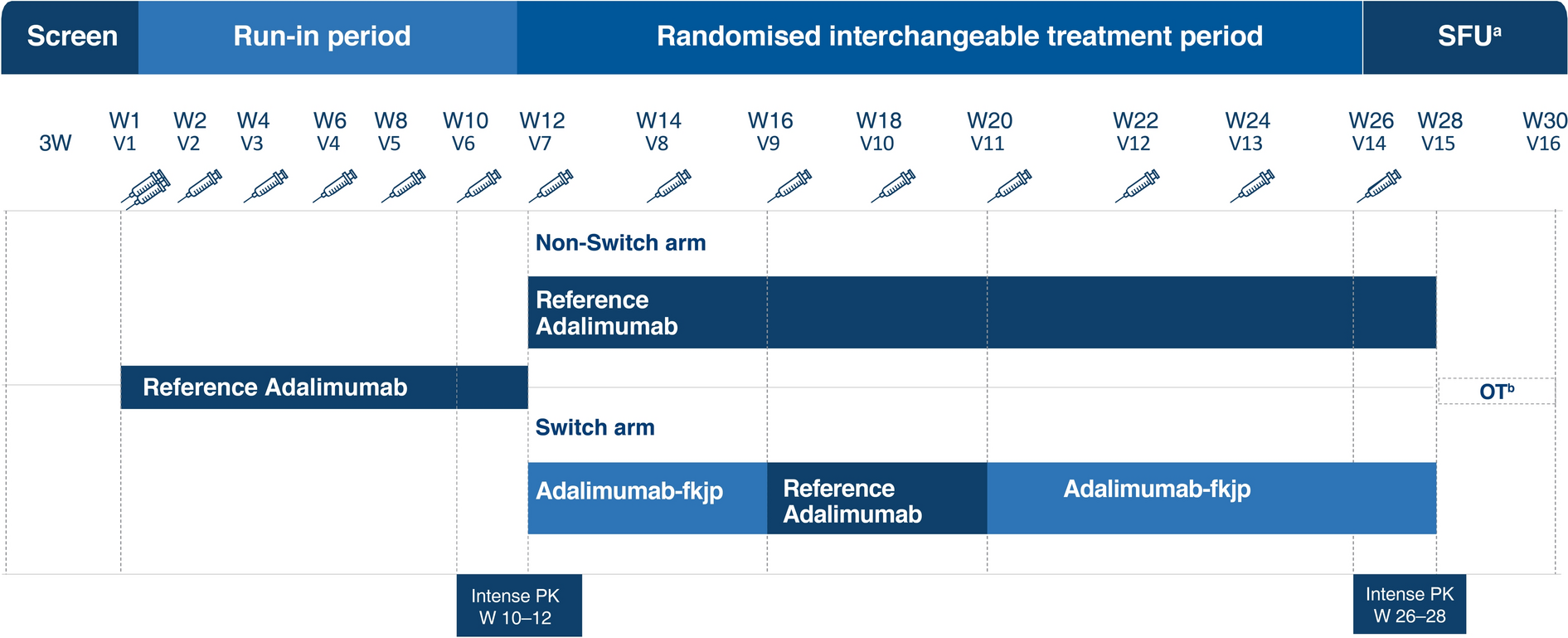
The design of the study is provided in Fig. 1. After a successful screening between Day 21 to Day 1, patients received reference adalimumab (80 mg, Week 1; 40 mg biweekly in Weeks 2–10). Patients achieving Psoriasis Area and Severity Index (PASI) 50 response at Week 12/Visit 7 were randomly assigned 1:1 to the following two groups for further treatment, non-switch arm: reference adalimumab (40 mg biweekly, Weeks 12–26), and switch arm: adalimumab-fkjp (40 mg, Weeks 12 and 14), reference adalimumab (40 mg, Weeks 16 and 18) and adalimumab-fkjp (40 mg, Weeks 20, 22, 24, 26). The last dose in the switch arm was at Week 26, and the follow-up safety assessments were carried out 4 weeks after the last dose, with the last efficacy and PK assessments carried out at Week 28. A total of three switches were performed for patients randomized to the switch group. The final switching duration provided a sufficient washout period for the reference adalimumab in the switching arm to reach low serum levels, which potentially maximized the generation and observation of the immunogenic response.
Fig. 1
Study design. Screening period (duration: up to 3 weeks): patients were enrolled in the study after successfully completing screening activities. Run-in period (duration: 11 weeks; Visits 1–6): patients received reference adalimumab [initial dose of 80 mg (2 × 40 mg); Week 1/Visit 1] administered SC, followed by 40 mg SC given every other week starting 1 week after the initial dose (last dose at Week 10/Visit 6). Randomized interchangeable treatment period (duration: 16 weeks; Visits 7–14): patients at the start of Week 12/Visit 7 were randomly assigned in a 1:1 ratio to either of the following groups and received 40 mg of the study drug every other week: non-switch arm: patients continued to receive reference adalimumab (Week 12/Visit 7 until Week 26/Visit 14, including both visits); switch arm: patients underwent repeated switches up to Week 26/Visit 14: adalimumab- fkjp at Week 12/Visit 7 and Week 14/Visit 8, reference adalimumab at Week 16/Visit 9 and Week 18/Visit 10, and adalimumab-fkjp at Week 20/Visit 11, Week 22/Visit 12, Week 24/Visit 13, and Week 26/Visit 14. aSFU safety follow-up assessment was carried out 4 weeks after the last dose. bOT optional treatment: additional doses of adalimumab at Week 28/Visit 14. W Week, V Visit, SC subcutaneously
Randomization was stratified based on Week 12/Visit 7 PASI response: ≥ PASI 50 to < PASI 75 or ≥ PASI 75, and was implemented by an automated Interactive Voice/Web Response system. As this was a blinded study, patients, investigators, sponsor personnel, and the study staff (except the unblinded staff receiving, handling, and administering the study medication at site), remained blinded to the randomized treatment assignments until after database lock.
PatientsPatients aged 18–75 years were eligible, upon providing consent, if they were diagnosed with moderate-to-severe chronic plaque psoriasis for ≥ 6 months at screening that had involved body surface area ≥ 10%, PASI ≥ 12, and static Physicians Global Assessment (sPGA) ≥ 3 (moderate) at screening and at baseline. Chronic plaque psoriasis is considered a sensitive indication to assess PK similarity in this interchangeability trial as adalimumab-fkjp and reference adalimumab are both administered as a monotherapy for this indication. The monotherapy setting results in a more homogeneous population with least interference from other commonly used concomitant medications (like methotrexate) for the evaluation of PK, safety, and immunogenicity.
For inclusion, the eligible patients had a stable disease for at least 2 months; were candidates for systemic therapy or phototherapy; had a previous failure, inadequate response, intolerance, or contraindication to at least one conventional anti-psoriatic systemic therapy, including methotrexate, cyclosporine, psoralen plus ultraviolet light A, and ultraviolet light B; and were willing to follow the contraception requirement, based on the childbearing potential. Patients were excluded if they were diagnosed with erythrodermic psoriasis, pustular psoriasis, guttate psoriasis, medication-induced psoriasis, other skin conditions, or other systemic autoimmune disorders or inflammatory diseases at the time of the screening, as these would have interfered with the effect and evaluations of the study treatment. Full inclusion and exclusion criteria are provided in Methods-S1 of the supplementary material. Table-S1 provides prior immunosuppressant treatments, including biologics, which were washed out before baseline (Week 1).
Endpoints and AssessmentsThe primary endpoint was to assess the adalimumab steady-state PK in the switch arm compared to the non-switch arm. The primary PK endpoints included AUCτ,26–28 (Area under the adalimumab concentration–time curve over the dosing interval of Weeks 26–28), and Cmax,26–28 (Maximum observed adalimumab concentration during the dosing interval of Weeks 26–28).
Secondary endpoints included: secondary PK endpoints, Tmax,26–28 and Cmin,26–28, during the intense PK sampling intervals between Weeks 26–28 and Ctrough at the scheduled PK sample time points; efficacy endpoints including the proportions of PASI 50, PASI 75, PASI 90, and PASI 100 responders and the proportions of sPGA success (clear or almost clear) at Week 28. Safety measures included type, incidence, severity, timing, seriousness, and relatedness of treatment-emergent adverse events (TEAEs) including injection site reactions (ISRs), hypersensitivity reactions, heart failure, malignancies, serious infections including tuberculosis, and laboratory test abnormalities. Immunogenicity was evaluated by observing the incidence of positive anti-drug antibody (ADA) and neutralizing antibody (NAb) response and ADA titers at Week 28.
Statistical AnalysisThe sample size was determined based on the primary PK endpoints. Equivalence between reference adalimumab and adalimumab-fkjp was established if the 90% confidence interval (CI) for the treatment difference was within 80.00–125.00% for both primary PK parameters. Based on this, a sample size of 370 patients, including a 10% run-in failure, ensured a power of 90% with 2 one-sided tests at a 5% significance level (per FDA requirements).
An ANCOVA model was used to analyze the AUCτ,26–28 and Cmax,26–28, which accounted for the (pre-randomization) covariables treatment (switch vs. non-switch arms), logarithm of PASI improvement (the ratio of PASI response at Week 12 and at Week 1), weight at screening period, and AUCτ,10–12 and Cmax,10–12. Least squares means (LSM) of the switch and non-switch arms were used to construct the point estimate for the ratio of means. The 90% CI was derived for the ratios of the means based on Fieller’s theorem. A conclusion of bioequivalence was made when the 90% CIs were within 80–125% for both AUCτ,26–28 and Cmax,26–28. The secondary PK endpoint, Cmin,26–28, was analyzed in the same way as the primary endpoints, with no requirement of a formal comparison to the bioequivalence range. The secondary PK endpoint, Tmax,26–28, was analyzed solely descriptively. CIs for the differences in the proportions of patients with PASI and sPGA responses in each arm were obtained using the Wald method. The safety analyses performed on the safety and run-in period populations were descriptive in nature, and immunogenicity to adalimumab was descriptively summarized by treatment group at Week 28. All statistical analyses were performed using Statistical Analysis System (SAS®), Version 9.4 or higher (SAS Institute, NC, USA).
Comments (0)