Study Design
This 3-month, exploratory, randomized, investigator-masked, active-controlled, parallel-group, multicenter study (EudraCT 2020-004836-93) was conducted in Europe. Four sites across Austria, Germany, and Greece were involved. The study was conducted in compliance with regulatory requirements provided by the following institutional review boards or independent ethics committees; the Faculty of Medicine at the Eberhard-Karls-University and the University Hospital Tübingen (reference number 193/2021), Ethics Committee of the University of Freiburg and the University of Vienna (reference number 1229/2021), Ethics Committee for the Province of Salzburg, and the Hellenic Republic Ministry of Health National Ethics Committee (reference number 34443/2021), in accordance with the protocol, Good Clinical Practice, The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use International guidelines, and the Declaration of Helsinki. All participants provided written informed consent prior to their participation.
Participants and Procedures
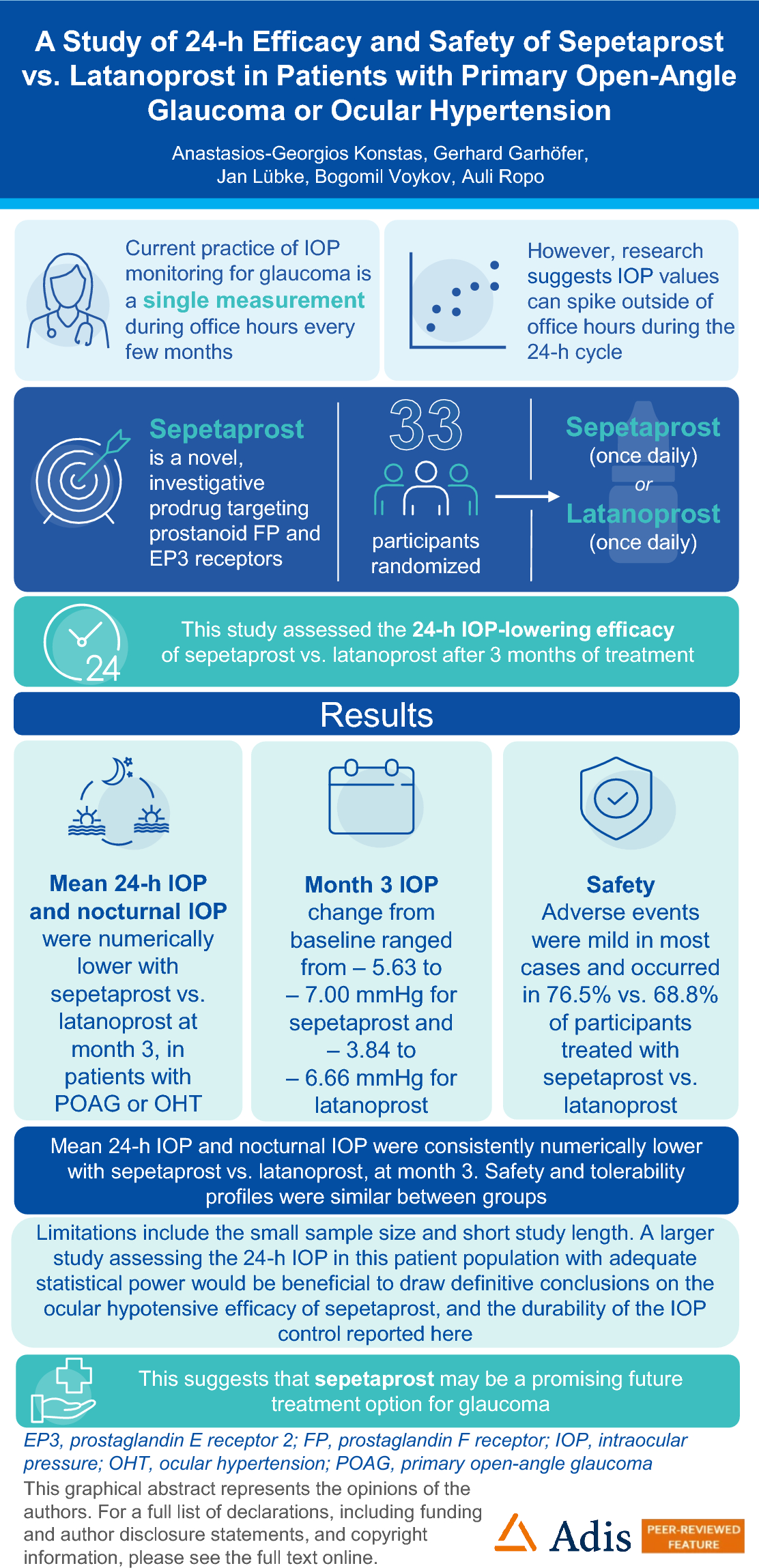
The study included a ≤ 35-day screening period (inclusive of a washout period of up to 28 + 7 days) and a 3-month treatment period where participants were randomized 1:1 to preservative-free sepetaprost ophthalmic solution 0.002% or preserved, branded latanoprost ophthalmic solution 0.005%. An overview of the study design is presented in Fig. S1, supplementary material.
At visit 1, participants were screened against the eligibility criteria; eligible participants entered the washout period and were instructed to discontinue use of all IOP-lowering medications. During the washout period, qualified participants who had discontinued IOP-lowering treatment were treated with a short-acting IOP-lowering agent (topical carbonic anhydrase inhibitor [CAI]) if deemed necessary by study investigators. The topical CAI was stopped 1 week before the eligibility visit (visit 2, day − 2).
Following the washout period, eligibility to enter the treatment period was determined at visit 2 based on the following inclusion criteria: participants ≥ 18 years of age with a diagnosis of POAG or OHT in both eyes or one eye with POAG and the other with OHT; best-corrected visual acuity (BCVA) of + 0.60 logarithm of the minimum angle of resolution (Snellen equivalent 20/80) or better in each eye; central corneal thickness ≥ 480 µm and ≤ 600 µm in each eye; anterior chamber angle grade ≥ 2 (Shaffer scale) in each eye; IOP of ≥ 22 mmHg in at least one eye, and ≤ 34 mmHg in both eyes at all time points of IOP measurements (08:00, 12:00, and 16:00) at visit 2.
At visits 1 and 2, participants with any of the following key exclusion criteria were not eligible to participate in the study: those who were pregnant, nursing, or planning a pregnancy; participation in other investigational drug (oral or topical therapy) or device clinical trials within 28 days prior to visit 2, and/or participation in other investigational drug (intravitreal injection therapy) clinical trials within 3 months prior to visit 2, or planning to participate in another investigational drug clinical trial that would have overlapped with the duration of this study; participants who could not safely discontinue use of ocular hypotensive medications during the washout period; presence of advanced glaucoma, corneal abnormality, severe external ocular disease, inflammation or infection of the eye, or history of iritis and/or uveitis or severe ocular trauma. The study eye was defined as the eye that met the pre-determined eligibility criteria at visit 2. If both eyes met the criteria, the eye with the higher MD IOP was designated as the study eye. If both eyes had equal IOP, the right eye was selected as the study eye.
Eligible participants were randomized to receive either preservative-free sepetaprost ophthalmic solution 0.002% or preserved latanoprost ophthalmic solution 0.005% every evening at 20:00 (± 60 min) in both eyes from visit 3 (baseline, day 1) to the evening prior to visit 6 (month 3). A permuted-block randomization was employed to randomize participants, and an Interactive Response Technology (Medidata Rave RTSM) system was used to generate the treatment assignments. The study was single-masked; investigators, examiners, and personnel involved in the conduct of the study were masked from study treatment.
During the 3-month treatment period, follow-up visits were scheduled at baseline (visit 3, day − 1), week 2 (visit 4), week 6 (visit 5), and month 3 (visit 6). A telephone follow-up visit was conducted at approximately 2 weeks following the last study drug administration to assess for adverse events (AEs). Baseline 24-h IOP was measured at 20:00 on day − 1, and at 24:00, 4:00, 8:00, 12:00, 16:00, and 20:00 on day 1. IOP was measured at 8:00 and 12:00 at week 2, and at 24:00, 4:00, 8:00, 12:00, 16:00, and 20:00 after the last dose given the previous night at 20:00 at week 6 and month 3. Peak and trough 24-h IOP and 24-h fluctuation (difference between the highest IOP reading minus the lowest IOP reading within the 24-h curve for each participant) in IOP were measured at day 1, week 6, and month 3.
Safety Analyses
The safety variables assessed included ocular and non-ocular AEs, and suspected adverse reactions (SARs; AEs assessed as related to the study drug by the investigators). At all scheduled visits, participants were questioned regarding potential AEs; investigators asked participants whether there had been a change in their general health, and direct questioning and examination was then performed as appropriate. AE information was also collected by telephone 2 weeks after the last study drug administration.
Outcomes
The primary endpoint was mean 24-h IOP in the study eye with sepetaprost or latanoprost measured at month 3. The secondary endpoints included mean 24-h IOP in the study eye with sepetaprost vs. latanoprost measured at week 6, mean diurnal IOP (8:00, 12:00, 16:00, and 20:00) and mean nocturnal IOP (24:00 and 4:00), IOP at week 6 and month 3, IOP at individual time points of the 24-h IOP curve at week 6 and month 3, peak and trough 24-h IOP and 24-h fluctuation in IOP at week 6 and month 3, IOP recorded at 36 h (8:00), 42 h (14:00), and 48 h (20:00) at month 3 + 1 day after the last dose of sepetaprost, vs. 12 h (8:00), 18 h (14:00), and 24 h (20:00) for latanoprost, and change from baseline in IOP at each time point at week 6 and month 3.
Safety endpoints included incidence of ocular and non-ocular AEs, and serious AEs (SAEs).
Statistical Methods
The sample size of this study was not based on any statistical power calculation. The sample size of 20 per treatment group was based on the exploratory nature of the study and the feasibility of conducting the trial. No formal statistical hypotheses testing was performed in this exploratory study; therefore, descriptive statistics were used to summarize all endpoints, including safety outcome measures. The 95% confidence intervals (CIs) were provided using a two-sample t test to calculate the difference between treatment groups. To account for differences in baseline IOP scores, an analysis of covariance (ANCOVA) was conducted, with baseline IOP included as a covariate.
The safety population included all randomized participants who received at least one dose of study drug. The safety analyses were performed on the safety population by actual treatment received.
The full analysis set (FAS) included all randomized participants who received at least one dose of study drug and who provided at least one post-baseline IOP measurement. Efficacy analyses were performed using the FAS or a subset of the FAS by planned treatment.




Comments (0)